
Research Articles:
Microbial Cell, Vol. 2, No. 4, pp. 126 - 135; doi: 10.15698/mic2015.04.196
Modeling human Coenzyme A synthase mutation in yeast reveals altered mitochondrial function, lipid content and iron metabolism

1 Department of Life Sciences, University of Parma, Parma, Italy.
2 Unit of Molecular Neurogenetics – Pierfranco and Luisa Mariani Center for the study of Mitochondrial Disorders in Children, IRCCS Foundation Neurological Institute “C. Besta”, Milan, Italy.
Keywords: Saccharomyces cerevisiae, yeast model, Coenzyme A, NBIA, COASY, mitochondria, iron accumulation, lipid content.
Abbreviations:
COASY - Coenzyme A synthase,
CoPAN - COASY protein-associated neurodegeneration,
COX - cytochrome c oxidase,
NCCR - NADH-cytochrome c oxidoreductase,
NBIA - neurodegeneration with brain iron accumulation,
PKAN - pantothenate kinase associated neurodegeneration.
Received originally: 05/12/2014 Received in revised form: 16/03/2015
Accepted: 22/03/2015
Published: 06/04/2015
Correspondence:
Paola Goffrini, Department of Life Sciences, University of Parma, Parma, Italy paola.goffrini@unipr.it
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Camilla Ceccatelli Berti, Cristina Dallabona, Mirca Lazzaretti, Sabrina Dusi, Elena Tosi, Valeria Tiranti, Paola Goffrini (2015). Modeling human Coenzyme A synthase mutation in yeast reveals altered mitochondrial function, lipid content and iron metabolism. Microbial Cell 2(4): 126-135.
Abstract
Mutations in nuclear genes associated with defective coenzyme A biosynthesis have been identified as responsible for some forms of neurodegeneration with brain iron accumulation (NBIA), namely PKAN and CoPAN. PKAN are defined by mutations in PANK2, encoding the pantothenate kinase 2 enzyme, that account for about 50% of cases of NBIA, whereas mutations in CoA synthase COASY have been recently reported as the second inborn error of CoA synthesis leading to CoPAN. As reported previously, yeast cells expressing the pathogenic mutation exhibited a temperature-sensitive growth defect in the absence of pantothenate and a reduced CoA content. Additional characterization revealed decreased oxygen consumption, reduced activities of mitochondrial respiratory complexes, higher iron content, increased sensitivity to oxidative stress and reduced amount of lipid droplets, thus partially recapitulating the phenotypes found in patients and establishing yeast as a potential model to clarify the pathogenesis underlying PKAN and CoPAN diseases.
INTRODUCTION
In all living cells Coenzyme A (CoA) is the major carrier of acetyl and acyl groups playing a central role in basic cellular functions such as lipids metabolism, Krebs cycle and aminoacid biosynthesis. CoA biosynthesis proceeds through a highly conserved pathway, involving five enzymatic steps: pantothenic acid (vitamin B5) phosphorylation, cysteine conjugation, decarboxylation, conjugation to an adenosyl group and phosphorylation.
–
Whereas in mammals the last two steps are catalyzed by Coenzyme A synthase (COASY), a mitochondrial bifunctional enzyme possessing both 4’-phospho-pantetheine adenylyltransferase (PPAT) and dephospho-CoA kinase (DPCK) activities [1][2], in other organisms, such as yeast, PPAT and DPCK activities reside in two different enzymes, Cab4 and Cab5, the products of the essential genes CAB4 and CAB5 [3] whose compartmentalization is not well understood.
–
Recently, it has been reported that dysfunctions of CoA biosynthetic pathway may play a role in the pathogenesis of neurodegeneration with brain iron accumulation (NBIA), a wide spectrum of clinically and genetically heterogeneous diseases characterized by progressive neurodegeneration and high iron content in specific brain region [4][5][6].
–
This concept is supported by the fact that mutations in PANK2, encoding the first enzyme in the CoA synthesis, approximately account for 50% of NBIA cases, classified as PKAN (Pantothenate Kinase Associated Neurodegeneration) [7][8]. Moreover COASY gene has been identified as a novel NBIA-associated gene and these NBIA cases have been termed CoPAN (COASY Protein-Associated Neurodegeneration)[9].
–
Neurodegenerative diseases are often characterized by mitochondrial dysfunctions, altered lipid metabolism and iron accumulation [10][11][12] and several evidences linking PKAN and CoPAN to these metabolic alterations have been reported [8][13][14][15][16].
–
The development of cellular and animal models is crucial for advancing our understanding of the pathophysiology of these diseases. In the last decade, the yeast Saccharomyces cerevisiae has been used as in vivo model system to gain insights into the molecular basis of mitochondrial pathologies and neurodegenerative disorders [17][18]. Despite their simplicity, yeast cells possess most of the basic cellular machinery including pathways required for energy metabolism and protein homeostasis. Moreover, many of the genes and biological systems that function in yeast iron homeostasis are conserved throughout eukaryotes [19].
–
To investigate if defective CoA metabolism could underlie a more general disequilibrium of lipid metabolism and mitochondrial dysfunctions and its relationship with brain iron accumulation, we have performed phenotypic and biochemical investigation in a recently developed yeast model expressing the pathogenic missense mutation COASYR499C found in NBIA patients [9].
–
The results obtained in this study showed that yeast mutant defective in CoA biosynthesis recapitulates the most important phenotypes found in patients and validated this system to model CoPAN in order to help elucidating important cellular and biochemical aspects of mitochondrial, lipid and iron homeostasis underpinning this disease.
RESULTS
Cellular localization of yeast Cab5 protein
Proteomics studies [20] and in silico analysis using the PSORT and MITOPROT programs [21][22], which allows the prediction of protein localization, suggest for Cab5 a mitochondrial localization. Moreover human CoA synthase is a mitochondrial enzyme and the human gene is able to complement the cab5Δ mutation. To confirm experimentally the mitochondrial localization of Cab5p, a carboxyl-terminal fusion of HA epitope to Cab5 was constructed. The cab5Δ lethal phenotype was rescued by the re-expression of the tagged wild type allele, indicating that the addition of HA did not disrupt targeting and function of the Cab5 protein. Equivalent amounts of mitochondrial pellet (M) and supernatant (PMS) fractions from cells expressing HA tagged Cab5 (Cab5-HA) were subjected to SDS-PAGE and Western blotting to identify the indicated protein. The great majority of Cab5-HA co-fractionated with the mitochondrial membrane protein porin, while only a small amount remained in the supernatant with the soluble cytoplasmic protein phosphoglycerate kinase (PGK), indicating that Cab5 behaves as a mitochondrial associated protein (Fig. 1A). We further investigated the mitochondrial localization of Cab5p by performing a protease protection assay of intact mitochondria. Cab5-HA exhibited a significant increase in proteinase K sensitivity treatment in respect to both porin, which is only partially exposed on the surface, and to the inner membrane protein Core1 (Fig. 1B). The mitochondria were then treated with 0.1 M Na2CO3, pH 11, and supernatant and pellet fractions were generated by centrifugation. As depicted in Fig. 1C the amount of Cab5 associated with mitochondria was significantly reduced but the amount of porin was not altered. Taken together these results suggest that Cab5 is an extrinsic outer membrane protein.
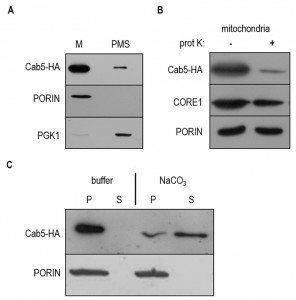 | FIGURE 1: Localization of Cab5p. (A) Equal amounts (20 mg) of the mitochondrial fraction (M) and post mitochondrial fraction (PMS) were resolved by SDS-PAGE and analyzed by immunoblotting with HA, PGK1 (cytosolic marker), PORIN (mitochondrial outer membrane marker) antibodies. (B) Mitochondria were treated for 60 min at 4°C with proteinase K (prot K) (1 mg/ml). The filter was incubated with anti-HA, anti-CORE1, and anti-PORIN antibodies. Core1 was used as an inner membrane protein control. (C) 150 µg of mitochondrial proteins were treated with TEGN buffer or TEGN and 0.1M NaCO3. The insoluble pellet (P) and supernatant (S) fractions were resolved by SDS-PAGE and analyzed by immunoblotting with HA and PORIN antibodies. |
Characterization of mitochondrial functions
We have previously demonstrated by HPLC analysis that in the strain expressing the human COASYR499C or the yeast cab5R146C mutant versions the level of CoA in mitochondria was reduced by 40% compared to wild-type [9]. Given that defective CoA biosynthesis could lead to a variety of metabolic defects we looked for evidence of mitochondrial dysfunction.
–
In order to reveal a possible respiratory growth defect, serial dilutions of the strains were spotted on mineral medium without pantothenate supplemented with either ethanol or glycerol, at 28°C. As shown in Fig. 2A the OXPHOS growth of the cab5Δ/COASYR499C mutant was partially affected compared to COASY wild type expressing strain. To confirm the growth delay we determined the cell
yield for each yeast strain grown on ethanol or glycerol. We observed that the OXPHOS growth of the mutant strain was 20% lower as compared to wild type (Fig. 2B).
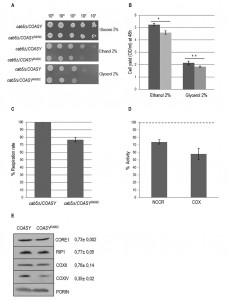 | FIGURE 2: Characterization of mitochondrial functions. (A) Oxidative growth phenotype. The strain W303-1B cab5Δ was transformed with a pYEX-BX plasmid carrying the wild-type COASY or the mutant allele COASYR499C. Equal amounts of serial dilutions of cells from exponentially grown cultures (105, 104, 103, 102, 101) were spotted onto mineral medium (40) plus 2% glucose, 2% ethanol or 2% glycerol without pantothenate. The growth was scored after 5 days of incubation at 28°C. (B) Cell yield. Cell yield was calculated by growing cells on liquid medium containing ethanol or glycerol and measuring the optical density at 600 nm after 48h of growth (COASY black columns and COASYR499C grey columns). Values are mean of three independent experiments. * P < 0.05; **P < 0.01 (unpaired two-tailed t-test). (C) Oxygen consumption rates. Respiration was measured in cells grown in mineral medium (40) plus 0.2% glucose and 2% galactose without pantothenate at 28°C. The values observed for the COASY mutant strain are reported as percentage of the respiration obtained in cells expressing the wild-type COASY gene. (D) NADH-cytochrome c oxidoreductase (NCCR) and cytochrome c oxidase (COX) activities were measured in mitochondria extracted from cells grown exponentially at 28°C in mineral medium (40) plus 0.2% glucose and 2% galactose without pantothenate. The values of the COASY mutant are expressed as percentage of the activities obtained in the wild type strain. (E) Steady state level of cIII and cIV subunits in cells carrying the wild-type COASY and the mutant allele. The filter was incubated with specific antibodies against Core1, Rip1, CoxII, CoxIV and Porin. The signals were normalized according to the control signal (porin) and taken as 1.00 the signal of the cab5∆/COASY (wild-type) strain. |
To further analyze the respiratory deficiency, oxygen consumption and activity of respiratory complexes were measured. Accordingly to the OXPHOS growth phenotype the oxygen consumption rate of the cab5Δ/COASYR499C was 25% less than that of cab5Δ/COASY (Fig. 2C). Likewise, the NADH-cytochrome c oxidoreductase (NCCR) and cytochrome c oxidase (COX) activities were reduced in the mutant strain respectively to 26% and 42% as compared to wild type (Fig. 2D). Accordingly, the steady state levels of complex III and IV subunits are decreased (Fig. 2E). Altogether these results indicate a mitochondrial dysfunction associated to the reduced CoA level.
–
Mutation in CoA synthase determines an increase of iron content and increased sensitivity to oxidative stress
NBIA disorders, PKAN and CoPAN included, are characterized by iron deposition in the brain but the mechanisms leading to iron overload and its pathophysiological role remain unclear. Since in yeast excessive iron accumulation in the mitochondria led to an increased sensitivity to this ion [23][24], we first evaluated the inhibition of cellular growth in the COASYR499C mutant strain by the addition of FeSO4 to the medium.
–
As depicted in Fig. 3A, the mutant strain showed a clear growth defect when compared to wild type strain, indirectly indicating a higher iron content.
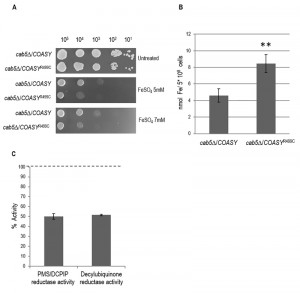 | FIGURE 3: Measurement of iron content. (A) Iron sensitivity. Equal amounts of serial dilutions of cells from exponentially grown cultures (105, 104, 103, 102, 101) were spotted onto 40 medium without pantothenate supplemented with 5 mM and 7 mM FeS04. The growth was scored after 5 days of incubation at 28°C. (B) Iron level. Cellular iron levels was quantified in cells grown up to early stationary phase in YNB glucose (0,6%) medium. **P < 0.01 (unpaired two-tailed t-test). (C) PMS/DCPIP (phenazyne methosulfate/dichlorophenolindophenol) reductase and decylubiquinone reductase activities were measured in mitochondria extracted from cells grown exponentially at 28°C in mineral medium (40) plus 0.2% glucose and 2% galactose without pantothenate. The values of the COASY mutant are expressed as percentage of the activities obtained in the wild type strain. |
We then performed a quantitative determination of cellular iron level by a colorimetric assay that relies on the formation of colored iron complexes with BPS after nitric acid digestion of yeast cells and gives results comparable with those with ICP-mass spectrometry [25][26]. The results obtained showed a two-fold increase in iron content in the COASYR499C mutant respect to the parental strain (Fig. 3B).
–
We then investigated whether the biosynthesis of the Fe-S cluster, a marker of mitochondrial functionality linked to iron metabolism, was affected by COASY deficiency. We determined the activity of succinate dehydrogenase (SDH), a mitochondrial Fe-S cluster enzyme. As shown in Fig. 3C, SDH activity was decreased by about 50%, in the mutant as compared to wild-type strain.
–
It is known that an excess of iron causes an altered oxidative status [24][27][28], another key feature of disease associated to CoA deficiency [14][29][30], which may be reflected in hypersensitivity to oxidative stress-induced cell death. To test this hypothesis COASYR499C mutant and control strain were exposed to H2O2 and cell viability was determined by both spot assay analysis (Fig. 4A) and by counting the formation of colonies (Fig 4B). At the highest H2O2 concentration tested (2 mM) wild type cells showed a viability of 10%, while mutant cells showed a viability of 2% (Fig. 4B) demonstrating that a COASY defect leads to oxidative stress susceptibility.
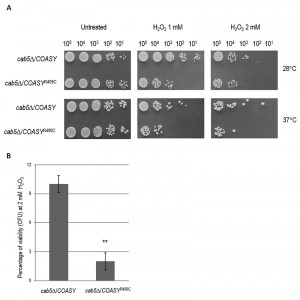 | FIGURE 4: Sensitivity to H2O2. The cells carrying the wild-type COASY or mutant allele were grown up to exponential phase and incubated for 4h at 28°C or 37°C with the addition of 1 mM and 2 mM H2O2. (A) After the treatment, equal amounts of serial dilutions of cells (105, 104, 103, 102, 101) were spotted onto YP media plus 2% glucose. The growth was scored after 2 days of incubation at 28°C or 37°C. (B) Viability of wild-type and mutant strains was measured by C.F.U counting after exposure of cell to 2 mM H2O2 for 4h.**P < 0.01 (unpaired two-tailed t-test). |
Evaluation of lipid droplets content
Acetyl-CoA is necessary for the production of neutral lipids, which serve as power reserve for the cell and are stored in lipid droplets. Since CoA is involved in the biosynthesis of fatty acids and having demonstrated that the mutant cab5Δ/COASYR499C shows a 40% reduction of coenzyme A, the content of intracellular lipid droplets in the mutant compared to the wild type was evaluated by fluorescence microscopy and fluorimetric analysis after incubation of the cells with the fluorescent lipophilic dye Nile Red [31]. As shown in Fig. 5A the content of lipid droplets is decreased in the mutant expressing the COASYR499C. In order to better quantify this reduction, the fluorescence of cells stained with Nile Red was measured using a fluorescence spectrometer. The measures, performed in triplicate, highlighted a reduction of lipid droplets of 25% in mutant strain compared to wild-type (Fig. 5B).
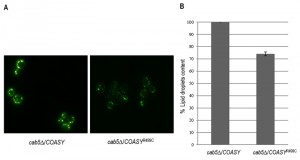 | FIGURE 5: Evaluation of lipid droplets content. The intracellular lipid droplets content has been detected by fluorescence microscopy (A) and fluorimetric analysis (B) after incubation of wild type and mutant cells with the fluorescent lipophilic dye Nile Red (4 μg/ml). The values corresponding to mutant COASY are expressed as percentage of the content obtained in the wild type strain. |
DISCUSSION
In all living organisms Coenzyme A (CoA) is an essential cofactor in cellular metabolism. CoA biosynthesis follows a highly conserved pathway, involving five enzymatic steps, which utilize pantothenate (vitamin B5), ATP, and cysteine. Mutations in nuclear genes directly involved in CoA biosynthetic pathway have been identified as responsible for some forms of NBIA, namely PKAN and CoPAN. PKAN is caused by mutations in PANK2, encoding the pantothenate kinase 2 enzyme, that account for about 50% of NBIA cases, whereas mutations in CoA synthase COASY have been recently reported as the second inborn error of CoA synthesis leading to CoPAN [9]. In PKAN and CoPAN brain iron accumulation is dramatic but its link with defective CoA synthesis is unknown.
–
Moreover, many neurodegenerative diseases, PKAN and CoPAN included, are characterized by mitochondrial dysfunctions, oxidative stress, altered lipid metabolism but again the complex relationships linking these factors in the context of disease conditions remain to be elucidated.
–
Previous attempts to understand the mechanism of PKAN using animal models have met with limited success. A mouse model of PKAN exhibits azoospermia but lacks any neurological phenotype [32]. A Drosophila model of PKAN does have a brain phenotype, but this involves the formation of vacuoles, not iron accumulation [33]. The identification and generation of other cellular model may allow a deeper characterization of COASY and PANK2 disease gene products, and the investigation of their pathophysiology in vivo. With this aim we developed and characterized a yeast model for CoPAN disease.
–
Although in yeast, differently from mammalian cells, the last two steps of CoA biosynthesis are catalyzed by two separate enzymes, namely the products of the essential genes CAB4 and CAB5, we have demonstrated that the lethality associated to deletion in CAB5 could be complemented by human COASY. This allowed us to study the human Arg499Cys substitution in yeast and to support the pathogenic role of this mutation associated to a reduced level of CoA [9].
–
The evaluation of the metabolic consequences of coenzyme A deficiency in yeast revealed mitochondrial dysfunctions; OXPHOS growth was affected and respiration rate significantly decreased. Accordingly, the activity of respiratory chain complexes and steady state levels of mitochondrial respiratory chain subunits were reduced. We also demonstrated that the growth of the mutant strain is not only strongly inhibited in the presence of iron but that the mutant strain showed iron accumulation. This result is consistent with the patient phenotype, with iron overload being a typical sign of PKAN and CoPAN. We have also found that cab5Δ/COASYR499C mutant was more sensitive to the ROS-inducing agent H2O2 indicating an increased oxidative stress that may contribute to the pathogenesis of these diseases. Accordingly, the activity of SDH, a marker of mitochondrial functionality linked to iron metabolism, was reduced in the COASY mutant.
–
Since Acetyl-CoA, one of the most important derivatives of CoA, is also required for the synthesis of fatty acids, we investigated the impact of reduced CoA level by measuring the lipid droplets content in mutant cells by fluorimetric analysis of Nile Red stained cells. Interestingly enough, lipid droplets content was 25% lower in mutant strain as compared to wild-type. The same altered lipid metabolism was also observed in mutant strain for phosphopantothenoylcysteine synthetase (PPCS), another essential enzyme in CoA biosynthetic pathway [34]. These results are in agreement with the hypothesis that low CoA perturbs lipid homeostasis; lipid deregulation was also observed in Drosophila CoA mutants and from global metabolic profiling studies in patient-derived fibroblasts [8][35]. The transcriptional analysis of key genes involved in lipid metabolism would help in elucidating the role of lipid metabolism in the pathology.
–
Altogether these results established yeast as an appropriate model to study the molecular mechanisms involved in CoA metabolism, and to understand the connection between iron management, mitochondrial function and lipid metabolism in neurodegeneration.
–
Several pathological phenotypes have been identified in the mutant COASY yeast strain thus representing ideal readouts for high throughput screening of chemical libraries as described by Couplan et al. [36]. This could allow the identification of new molecules, the first step to set up future therapeutic experimental approaches.
MATERIALS AND METHODS
Yeast strains, media and vectors
Yeast strains used in this study were W303-1B cab5Δ (MATα; cab5::KanMx4 ade2-1 leu2-3,112 ura3-1 his3-22,15 trp1-1 can1-100) carrying pYEX-BX-COASY or pYEX-BX COASYR499C plasmid [9]. For localization studies we used the strain BY4741 (MATa; his3∆1 leu2∆0 met15∆0 ura3∆0) transformed with pFL38-Cab5-HA plasmid.
–
Cells were cultured in mineral medium (40) supplemented with appropriate amino acids and bases for auxotrophy as previously described [37]. To obtain medium lacking pantothenate (40-Pan) a mixture of vitamins without pantothenate was prepared. Yeast cells were transformed by the standard lithium acetate method [38] and cultured in YNB synthetic defined media (For-MediumTM, UK) supplemented with 1 g/l of drop-out powder [39] containing all amino acids and bases, except those required for plasmid selection. Various carbon sources (Carlo Erba Reagents, Italy) were added at the indicated concentration. Media were solidified with 20 g/l agar (For-Medium™). Strains were incubated at 28 or 37°C as indicated.
–
Plasmid pFL38-Cab5-HA was obtained by PCR overlap technique [40]. In the first set of PCR reactions, the CAB5 region was obtained using the forward primer CAB5Fw-GGGGGGATCCCCATTGCTTAGAATGGGCGG and the following reverse tag primer CAB5HARv ATCAACCTTATACAGCGTAATCTG-GAACATCGTATGGGTACGCTGAAGACTTTTTATTTTG where hemagglutinin (HA) tag sequence is indicated in bold. The second CAB5 region was obtained using the forward tag primers CAB5HATERFw complementary to CAB5HARv , and the reverse primer CAB5Rv-CCGCGGTACCGAGAACCCATAGAATTCGAC. The final product was obtained using the overlapping PCR fragments as template with CAB5Fw and CAB5Rv as external primers. The product was then digested with BamHI and KpnI and cloned in BamHI/KpnI digested pFL38 centromeric plasmid [41].
–
Respiration measurement, biochemical assays and immunoblot analysis of respiratory chain subunits.
Respiratory activity measurement was performed on whole cells at 30°C using a Clark-type oxygen electrode (Oxygraph System, Hansatech Instruments, England) with 1 ml of air-saturated respiration buffer (0.1 M phthalate–KOH, pH 5.0). The reaction started by addition of 20 mg of wet-weight cells [42].
–
Complex II (SDH), NADH-cytochrome c oxidoreductase (NCCR) and complex IV specific activities were measured spectrophotometrically as previously described [42][43][44][45] on a mitochondrial enriched fraction prepared according to Soto et al. [46]. Protein concentration was determined by the method of Bradford using the BioRad protein assay following manufacturer’s instructions [47]. For SDS-PAGE, 20 μg of mitochondrial proteins were separated on 12% polyacrylamide gels and electroblotted onto a nitrocellulose membrane. The subunits of mitochondrial respiratory complexes were immunovisualized by specific antibodies. The sources of primary antibodies are indicated: anti-Core1 (a kind gift from Prof. Antoni Barrientos), anti-Rip1 (a kind gift from Prof. Alexander Tzagoloff), anti CoxIIp (Abcam Mitoscience), anti-CoxIV (Abcam Mitoscience) and anti-Porin (Abcam Mitoscience). Quantification of protein bands was performed using Multi Analyst software. The signals were normalized according to the control signal (α-Porin) and the signal of the cab5∆/COASY (wild-type) strain was set as 1.00.
–
Intact mitochondria isolation, subcellular localization experiments and membrane association
Intact mitochondria were isolated from BY4741 strain transformed with a plasmid expressing Cab5-HA under the native CAB5 promoter after cell wall removal by Zymoliase20T digestion (Nacalai Tesque) and cell disruption with a glass-teflon potter homogenizer [48]. Whole cell extract was centrifuged at 12,000 g for 30 min to yield the mitochondrial pellet (M) and post-mitochondria supernatant (PMS). These fractions were analyzed by immunoblotting with the indicated antibodies (Porin: mitochondrial marker; PGK cytoplasmic marker (Abcam Mitoscience)). The Cab5 protein was immunovisualized using an anti-HA (Roche) specific antibody. Proteinase K protection assay for sub-mitochondrial localization study was performed as previously described [48][49]. Briefly, 200 μg of mitochondrial proteins were kept in 20 mM HEPES pH 7.4, 0,6 M sorbitol in the presence or absence of proteinase K (1 mg/ml) for 60 minutes on ice. 0,1 M PMSF (phenylmethylsulfonyl fluoride) was added to stop the reaction. The protein pellets were washed once with 20 mM HEPES pH 7.4 plus 0,6 M sorbitol, and suspended in SDS-PAGE sample buffer.
–
A modified version of the membrane association experiments of Trott and Morano [50] was utilized to determine the resistance of Cab5p to sodium carbonate (pH 11.5) treatment. Equal amounts (150 μg) of the mitochondrial fraction was resuspended in TEGN (20 mM Tris-HCl [pH 7.9], 0.5 mM EDTA, 10% glycerol, 50 mM NaCl) or TEGN and with 0.1 M NaCO3 for 30 min on ice. The samples were subsequently centrifuged at 17,000 g at 4°C to obtain soluble and membrane fractions. The fractions obtained in all type of extraction were separated by SDS-PAGE and probed with anti-HA and anti-PORIN antibodies.
–
Measurement of iron content
The iron content was determined by a colorimetric assay, essentially as described before [25][51]. 5×108 cells were washed twice with H2O, resuspended in 0.5 ml of 3% nitric acid and incubated over night at 95°C. After incubation, samples were centrifuged at 12,000 rpm for 5 min and the supernatant (400 µl) was mixed with 160 µl of 38 mg sodium L-ascorbate ml– 1 (SIGMA), 320 µl of 1.7 mg BPS ml-1 (ACROS ORGANICS), and 126 µl of ammonium acetate (SIGMA) (saturated solution diluted 1:3). Non-specific absorbance was measured at 680 nm and subtracted from the specific absorbance of the iron-BPS complex (535 nm). Iron was quantified by reference to a standard curve using iron sulfate performed as in Tamarit et al. [25].
–
H2O2 sensitivity
To determine the sensitivity to oxygen peroxide, cells growing exponentially were exposed to 1 and 2 mM H2O2 at 28°C or 37°C for 4 h. Cell viability was determined by spotting equal amounts of serial dilutions of cells (105, 104, 103, 102, 101) onto YP plates (1% yeast extract, 2% peptone ForMediumTM) supplemented with 2% glucose (YPD). Plates were incubated at 28°C or 37°C for two days. To better quantify H2O2 sensitivity cell survival was determined by counting the formation of colonies after the treatment.
–
Lipid droplets content: fluorescence microscopy and fluorimetric analysis
Intracellular lipid droplets were detected using the fluorescent lipophilic dye Nile Red (9-diethylamino-5H-benzo[α]phenoxazine-5-one 3 SIGMA-ALDRICH) by fluorescence microscopy and fluorimetric analysis [31][52][53]. Wild type and cab5∆/COASYR499C strains were grown to mid-log phase in mineral medium (40) containing Yeast Extract (1,5 g/L). To 250 μl of the cultures, adjusted to 1 OD, 10 μl of the stock solution of Nile red [100 μg/ml] were added in order to obtain a final concentration of 4 µg/ml of dye. Fluorescence of the stained cells were obtained with a Leica DM2000 microscope using x 100 magnification and captured using a Leica DFC310FX digital camera with Leica’s Imaging Software (Leica Application Suite-LASAF 3.7.0, Leica Microsystem).
–
To quantify the fluorescence we used the fluorescence spectrometer Tecan SPECTRA Fluor Plus using the software XFLUOR4 V4.51 (excitation at 535 nm and emission at 595 nm). Aliquots of 100 μl of cells stained with Nile red were transferred into 96-well microplates in 4 replicates. For each strain a negative control was performed in which the dye was omitted in order to exclude a possible auto fluorescence of samples. The evaluation of the fluorescence was repeated at 5-minute intervals in a time interval of 20 minutes [53].
References
- A. Zhyvoloup, I. Nemazanyy, A. Babich, G. Panasyuk, N. Pobigailo, M. Vudmaska, V. Naidenov, O. Kukharenko, S. Palchevskii, L. Savinska, G. Ovcharenko, F. Verdier, T. Valovka, T. Fenton, H. Rebholz, M. Wang, P. Shepherd, G. Matsuka, V. Filonenko, and I.T. Gout, "Molecular cloning of CoA Synthase. The missing link in CoA biosynthesis.", The Journal of biological chemistry, 2002. http://www.ncbi.nlm.nih.gov/pubmed/11980892
- A. Zhyvoloup, I. Nemazanyy, G. Panasyuk, T. Valovka, T. Fenton, H. Rebholz, M. Wang, R. Foxon, V. Lyzogubov, V. Usenko, R. Kyyamova, O. Gorbenko, G. Matsuka, V. Filonenko, and I.T. Gout, "Subcellular Localization and Regulation of Coenzyme A Synthase", Journal of Biological Chemistry, vol. 278, pp. 50316-50321, 2003. http://dx.doi.org/10.1074/jbc.M307763200
- J. Olzhausen, S. Schübbe, and H. Schüller, "Genetic analysis of coenzyme A biosynthesis in the yeast Saccharomyces cerevisiae: identification of a conditional mutation in the pantothenate kinase gene CAB1", Current Genetics, vol. 55, pp. 163-173, 2009. http://dx.doi.org/10.1007/s00294-009-0234-1
- A. Gregory, and S.J. Hayflick, "Neurodegeneration with brain iron accumulation.", Folia neuropathologica, 2005. http://www.ncbi.nlm.nih.gov/pubmed/16416393
- D. Brunetti, S. Dusi, M. Morbin, A. Uggetti, F. Moda, I. D'Amato, C. Giordano, G. d'Amati, A. Cozzi, S. Levi, S. Hayflick, and V. Tiranti, "Pantothenate kinase-associated neurodegeneration: altered mitochondria membrane potential and defective respiration in Pank2 knock-out mouse model", Human Molecular Genetics, vol. 21, pp. 5294-5305, 2012. http://dx.doi.org/10.1093/hmg/dds380
- S. Hayflick, "Defective pantothenate metabolism and neurodegeneration", Biochemical Society Transactions, vol. 42, pp. 1063-1068, 2014. http://dx.doi.org/10.1042/BST20140098
- M.B. Hartig, K. Hörtnagel, B. Garavaglia, G. Zorzi, T. Kmiec, T. Klopstock, K. Rostasy, M. Svetel, V.S. Kostic, M. Schuelke, E. Botz, A. Weindl, I. Novakovic, N. Nardocci, H. Prokisch, and T. Meitinger, "Genotypic and phenotypic spectrum of PANK2 mutations in patients with neurodegeneration with brain iron accumulation.", Annals of neurology, 2006. http://www.ncbi.nlm.nih.gov/pubmed/16437574
- V. Leoni, L. Strittmatter, G. Zorzi, F. Zibordi, S. Dusi, B. Garavaglia, P. Venco, C. Caccia, A.L. Souza, A. Deik, C.B. Clish, M. Rimoldi, E. Ciusani, E. Bertini, N. Nardocci, V.K. Mootha, and V. Tiranti, "Metabolic consequences of mitochondrial coenzyme A deficiency in patients with PANK2 mutations", Molecular Genetics and Metabolism, vol. 105, pp. 463-471, 2012. http://dx.doi.org/10.1016/j.ymgme.2011.12.005
- S. Dusi, L. Valletta, T. Haack, Y. Tsuchiya, P. Venco, S. Pasqualato, P. Goffrini, M. Tigano, N. Demchenko, T. Wieland, T. Schwarzmayr, T. Strom, F. Invernizzi, B. Garavaglia, A. Gregory, L. Sanford, J. Hamada, C. Bettencourt, H. Houlden, L. Chiapparini, G. Zorzi, M. Kurian, N. Nardocci, H. Prokisch, S. Hayflick, I. Gout, and V. Tiranti, "Exome Sequence Reveals Mutations in CoA Synthase as a Cause of Neurodegeneration with Brain Iron Accumulation", The American Journal of Human Genetics, vol. 94, pp. 11-22, 2014. http://dx.doi.org/10.1016/j.ajhg.2013.11.008
- G.R. Campbell, J.T. Worrall, and D.J. Mahad, "The central role of mitochondria in axonal degeneration in multiple sclerosis.", Multiple sclerosis (Houndmills, Basingstoke, England), 2014. http://www.ncbi.nlm.nih.gov/pubmed/25122475
- G.M. Palomo, and G. Manfredi, "Exploring new pathways of neurodegeneration in ALS: The role of mitochondria quality control", Brain Research, vol. 1607, pp. 36-46, 2015. http://dx.doi.org/10.1016/j.brainres.2014.09.065
- P.J. Urrutia, N.P. Mena, and M.T. Núñez, "The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders", Frontiers in Pharmacology, vol. 5, 2014. http://dx.doi.org/10.3389/fphar.2014.00038
- P.T. Kotzbauer, A.C. Truax, J.Q. Trojanowski, and V.M. Lee, "Altered neuronal mitochondrial coenzyme A synthesis in neurodegeneration with brain iron accumulation caused by abnormal processing, stability, and catalytic activity of mutant pantothenate kinase 2.", The Journal of neuroscience : the official journal of the Society for Neuroscience, 2005. http://www.ncbi.nlm.nih.gov/pubmed/15659606
- A. Campanella, D. Privitera, M. Guaraldo, E. Rovelli, C. Barzaghi, B. Garavaglia, P. Santambrogio, A. Cozzi, and S. Levi, "Skin fibroblasts from pantothenate kinase-associated neurodegeneration patients show altered cellular oxidative status and have defective iron-handling properties", Human Molecular Genetics, vol. 21, pp. 4049-4059, 2012. http://dx.doi.org/10.1093/hmg/dds229
- C. Colombelli, M. Aoun, and V. Tiranti, "Defective lipid metabolism in neurodegeneration with brain iron accumulation (NBIA) syndromes: not only a matter of iron", Journal of Inherited Metabolic Disease, vol. 38, pp. 123-136, 2014. http://dx.doi.org/10.1007/s10545-014-9770-z
- S. Levi, and D. Finazzi, "Neurodegeneration with brain iron accumulation: update on pathogenic mechanisms", Frontiers in Pharmacology, vol. 5, 2014. http://dx.doi.org/10.3389/fphar.2014.00099
- T. Rinaldi, C. Dallabona, I. Ferrero, L. Frontali, and M. Bolotin-Fukuhara, "Mitochondrial diseases and the role of the yeast models", FEMS Yeast Research, vol. 10, pp. 1006-1022, 2010. http://dx.doi.org/10.1111/j.1567-1364.2010.00685.x
- S. Tenreiro, M.C. Munder, S. Alberti, and T.F. Outeiro, "Harnessing the power of yeast to unravel the molecular basis of neurodegeneration", Journal of Neurochemistry, vol. 127, pp. 438-452, 2013. http://dx.doi.org/10.1111/jnc.12271
- M.R. Bleackley, and R.T.A. MacGillivray, "Transition metal homeostasis: from yeast to human disease", BioMetals, vol. 24, pp. 785-809, 2011. http://dx.doi.org/10.1007/s10534-011-9451-4
- J. Reinders, R.P. Zahedi, N. Pfanner, C. Meisinger, and A. Sickmann, "Toward the complete yeast mitochondrial proteome: multidimensional separation techniques for mitochondrial proteomics.", Journal of proteome research, 2006. http://www.ncbi.nlm.nih.gov/pubmed/16823961
- E.C. Uberbacher, and R.J. Mural, "Locating protein-coding regions in human DNA sequences by a multiple sensor-neural network approach.", Proceedings of the National Academy of Sciences of the United States of America, 1991. http://www.ncbi.nlm.nih.gov/pubmed/1763041
- M.G. Claros, and P. Vincens, "Computational method to predict mitochondrially imported proteins and their targeting sequences.", European journal of biochemistry, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8944766
- F. Foury, and O. Cazzalini, "Deletion of the yeast homologue of the human gene associated with Friedreich's ataxia elicits iron accumulation in mitochondria.", FEBS letters, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9271239
- V.A. Patil, J.L. Fox, V.M. Gohil, D.R. Winge, and M.L. Greenberg, "Loss of Cardiolipin Leads to Perturbation of Mitochondrial and Cellular Iron Homeostasis", Journal of Biological Chemistry, vol. 288, pp. 1696-1705, 2013. http://dx.doi.org/10.1074/jbc.M112.428938
- J. Tamarit, V. Irazusta, A. Moreno-Cermeño, and J. Ros, "Colorimetric assay for the quantitation of iron in yeast.", Analytical biochemistry, 2005. http://www.ncbi.nlm.nih.gov/pubmed/16403430
- S. Molik, R. Lill, and U. Mühlenhoff, "Methods for studying iron metabolism in yeast mitochondria.", Methods in cell biology, 2007. http://www.ncbi.nlm.nih.gov/pubmed/17445699
- B. Schilke, C. Voisine, H. Beinert, and E. Craig, "Evidence for a conserved system for iron metabolism in the mitochondria of Saccharomyces cerevisiae.", Proceedings of the National Academy of Sciences of the United States of America, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10468587
- U. Mühlenhoff, N. Richhardt, M. Ristow, G. Kispal, and R. Lill, "The yeast frataxin homolog Yfh1p plays a specific role in the maturation of cellular Fe/S proteins.", Human molecular genetics, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12165564
- M. Wu, H. Liu, W. Chen, Y. Fujimoto, and J. Liu, "Hepatic Expression of Long‐Chain Acyl‐CoA Synthetase 3 is Upregulated in Hyperlipidemic Hamsters", Lipids, vol. 44, 2009. http://dx.doi.org/10.1007/s11745-009-3341-3
- A. Rana, E. Seinen, K. Siudeja, R. Muntendam, B. Srinivasan, J.J. van der Want, S. Hayflick, D. Reijngoud, O. Kayser, and O.C.M. Sibon, "Pantethine rescues a Drosophila model for pantothenate kinase–associated neurodegeneration", Proceedings of the National Academy of Sciences, vol. 107, pp. 6988-6993, 2010. http://dx.doi.org/10.1073/pnas.0912105107
- M. Hirade, "[Relationship between periodontal disease and citric acid. II. The effect of scaling and brushing on the citric acid contents in pocket exudate and whole saliva (author's transl)].", Nihon Shishubyo Gakkai kaishi, 1978. http://www.ncbi.nlm.nih.gov/pubmed/397290
- Y. Kuo, J.L. Duncan, S.K. Westaway, H. Yang, G. Nune, E.Y. Xu, S.J. Hayflick, and J. Gitschier, "Deficiency of pantothenate kinase 2 (Pank2) in mice leads to retinal degeneration and azoospermia.", Human molecular genetics, 2004. http://www.ncbi.nlm.nih.gov/pubmed/15525657
- Y. Yang, Z. Wu, Y.M. Kuo, and B. Zhou, "Dietary rescue of fumble--a Drosophila model for pantothenate-kinase-associated neurodegeneration.", Journal of inherited metabolic disease, 2005. http://www.ncbi.nlm.nih.gov/pubmed/16435199
- "Open Biology", . http://dx.doi.org/10.1098/rsob
- F. Bosveld, A. Rana, P.E. van der Wouden, W. Lemstra, M. Ritsema, H.H. Kampinga, and O.C.M. Sibon, "De novo CoA biosynthesis is required to maintain DNA integrity during development of the Drosophila nervous system", Human Molecular Genetics, vol. 17, pp. 2058-2069, 2008. http://dx.doi.org/10.1093/hmg/ddn105
- E. Couplan, R.S. Aiyar, R. Kucharczyk, A. Kabala, N. Ezkurdia, J. Gagneur, R.P. St. Onge, B. Salin, F. Soubigou, M. Le Cann, L.M. Steinmetz, J. di Rago, and M. Blondel, "A yeast-based assay identifies drugs active against human mitochondrial disorders", Proceedings of the National Academy of Sciences, vol. 108, pp. 11989-11994, 2011. http://dx.doi.org/10.1073/pnas.1101478108
- G.E. Magni, and R.C. Von Borstel, "Different Rates of Spontaneous Mutation during Mitosis and Meiosis in Yeast.", Genetics, 1962. http://www.ncbi.nlm.nih.gov/pubmed/17248123
- R.H. Schiestl, and R.D. Gietz, "High efficiency transformation of intact yeast cells using single stranded nucleic acids as a carrier.", Current genetics, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2692852
- C. Kaiser, S. Michaelis, and A. Mitchell, "Methods in Yeast Genetics.", Cold Spring Harbor Laboratory Press., 1994.
- S.N. Ho, H.D. Hunt, R.M. Horton, J.K. Pullen, and L.R. Pease, "Site-directed mutagenesis by overlap extension using the polymerase chain reaction.", Gene, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2744487
- N. Bonneaud, O. Ozier-Kalogeropoulos, G.Y. Li, M. Labouesse, L. Minvielle-Sebastia, and F. Lacroute, "A family of low and high copy replicative, integrative and single-stranded S. cerevisiae/E. coli shuttle vectors.", Yeast (Chichester, England), 1991. http://www.ncbi.nlm.nih.gov/pubmed/1767589
- P. Goffrini, T. Ercolino, E. Panizza, V. Giache, L. Cavone, A. Chiarugi, V. Dima, I. Ferrero, and M. Mannelli, "Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor", Human Molecular Genetics, vol. 18, pp. 1860-1868, 2009. http://dx.doi.org/10.1093/hmg/ddp102
- F. Fontanesi, I.C. Soto, and A. Barrientos, "Cytochrome c oxidase biogenesis: New levels of regulation", IUBMB Life, vol. 60, pp. 557-568, 2008. http://dx.doi.org/10.1002/iub.86
- A. Barrientos, F. Fontanesi, and F. Díaz, "Evaluation of the Mitochondrial Respiratory Chain and Oxidative Phosphorylation System Using Polarography and Spectrophotometric Enzyme Assays", Current Protocols in Human Genetics, vol. 63, 2009. http://dx.doi.org/10.1002/0471142905.hg1903s63
- D. Jarreta, J. Orús, A. Barrientos, O. Miró, E. Roig, M. Heras, C.T. Moraes, F. Cardellach, and J. Casademont, "Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy.", Cardiovascular research, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10728411
- I.C. Soto, F. Fontanesi, M. Valledor, D. Horn, R. Singh, and A. Barrientos, "Synthesis of cytochrome c oxidase subunit 1 is translationally downregulated in the absence of functional F1F0-ATP synthase", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1793, pp. 1776-1786, 2009. http://dx.doi.org/10.1016/j.bbamcr.2009.09.002
- M.M. Bradford, "A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding.", Analytical biochemistry, 1976. http://www.ncbi.nlm.nih.gov/pubmed/942051
- B.S. Glick, and L.A. Pon, "Isolation of highly purified mitochondria from Saccharomyces cerevisiae.", Methods in enzymology, 1995. http://www.ncbi.nlm.nih.gov/pubmed/8592446
- K. Diekert, A.I. de Kroon, G. Kispal, and R. Lill, "Isolation and subfractionation of mitochondria from the yeast Saccharomyces cerevisiae.", Methods in cell biology, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11381604
- A. Trott, and K.A. Morano, "SYM1 is the stress-induced Saccharomyces cerevisiae ortholog of the mammalian kidney disease gene Mpv17 and is required for ethanol metabolism and tolerance during heat shock.", Eukaryotic cell, 2004. http://www.ncbi.nlm.nih.gov/pubmed/15189984
- T. Almeida, M. Marques, D. Mojzita, M.A. Amorim, R.D. Silva, B. Almeida, P. Rodrigues, P. Ludovico, S. Hohmann, P. Moradas-Ferreira, M. Côrte-Real, and V. Costa, "Isc1p plays a key role in hydrogen peroxide resistance and chronological lifespan through modulation of iron levels and apoptosis.", Molecular biology of the cell, 2007. http://www.ncbi.nlm.nih.gov/pubmed/18162582
- K. Kimura, M. Yamaoka, and Y. Kamisaka, "Rapid estimation of lipids in oleaginous fungi and yeasts using Nile red fluorescence.", Journal of microbiological methods, 2004. http://www.ncbi.nlm.nih.gov/pubmed/14967224
- I. Sitepu, L. Ignatia, A. Franz, D. Wong, S. Faulina, M. Tsui, A. Kanti, and K. Boundy-Mills, "An improved high-throughput Nile red fluorescence assay for estimating intracellular lipids in a variety of yeast species", Journal of Microbiological Methods, vol. 91, pp. 321-328, 2012. http://dx.doi.org/10.1016/j.mimet.2012.09.001
ACKNOWLEDGMENTS
The financial support of Telethon GGP11011, Telethon GGP11088 and Fondazione CARIPLO 2011/0526 are gratefully acknowledged. This work was supported by TIRCON project of the European Commission’s seventh Framework Programme (FP7/2007-2013, HEALTH-F2-2011, grant agreement no. 277984). We wish to thank Antonietta Cirasolo (University of Parma) for the skillful technical assistance.
COPYRIGHT
© 2015

Modeling human Coenzyme A synthase mutation in yeast reveals altered mitochondrial function, lipid content and iron metabolism by Camilla Ceccatelli Berti et al. is licensed under a Creative Commons Attribution 4.0 International License.








