
Reviews:
Microbial Cell, Vol. 4, No. 11, pp. 368 - 375; doi: 10.15698/mic2017.11.597
The integrated stress response in budding yeast lifespan extension

1 Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, 1300 York Avenue, New York, NY 10065.
2 Orentreich Foundation for the Advancement of Science, Cold Spring, NY.
Keywords: replicative lifespan, chronological lifespan, budding yeast, integrated stress response, autophagy.
Abbreviations:
CLS - chronological lifespan,
ISR - Integrated Stress Response,
RLS - replicative lifespan,
uORFs - upstream open reading frames,
UPR - unfolded protein response.
Received originally: 11/08/2017 Received in revised form: 04/10/2017
Accepted: 05/10/2017
Published: 24/10/2017
Correspondence:
Jessica K. Tyler, Department of Pathology and Laboratory Medicine, Weill Cornell Medicine, 1300 York Avenue, New York, NY 10065; jet2021@med.cornell.edu
Conflict of interest statement: The authors declare that they have no competing interests.
Please cite this article as: Spike D.L. Postnikoff, Jay E. Johnson and Jessica K. Tyler (2017). The integrated stress response in budding yeast lifespan extension. Microbial Cell 4(11): 368-375. doi: 10.15698/mic2017.11.597
Abstract
Aging is a complex, multi-factorial biological process shared by all living organisms. It is manifested by a gradual accumulation of molecular alterations that lead to the decline of normal physiological functions in a time-dependent fashion. The ultimate goal of aging research is to develop therapeutic means to extend human lifespan, while reducing susceptibility to many age-related diseases including cancer, as well as metabolic, cardiovascular and neurodegenerative disorders. However, this first requires elucidation of the causes of aging, which has been greatly facilitated by the use of model organisms. In particular, the budding yeast Saccharomyces cerevisiae has been invaluable in the identification of conserved molecular and cellular determinants of aging and for the development of approaches to manipulate these aging determinants to extend lifespan. Strikingly, where examined, virtually all means to experimentally extend lifespan result in the induction of cellular stress responses. This review describes growing evidence in yeast that activation of the integrated stress response contributes significantly to lifespan extension. These findings demonstrate that yeast remains a powerful model system for elucidating conserved mechanisms to achieve lifespan extension that are likely to drive therapeutic approaches to extend human lifespan and healthspan.
INTRODUCTION TO YEAST AGING
The intent of lifespan-extending regimens is to prevent and treat the major diseases of society. However aging studies in humans and mammals are hampered by their very long lifespan. Thus, short-lived organisms have gained popularity as experimental models for aging studies. In particular, budding yeast is a highly informative model for aging studies with its short lifespan, genetic tools, and fully sequenced genome [1][2]. Though unicellular, yeast has been a remarkable experimental model to identify and characterize evolutionarily conserved basic biological processes, including aging. Accordingly, research in budding yeast continues to uncover the conserved pathways that cause aging in all eukaryotes [3], leading to the identification of genes and interventions that can mediate lifespan extension. For example, potential anti-aging drugs such as rapamycin, resveratrol, and spermidine, and promising longevity factors (e.g. Sir2, Tor1, Sch9, Ras) were first identified and characterized in yeast before they were adopted and confirmed to work in metazoans [4].
–
Budding yeast provides two separate, but partially overlapping paradigms for aging studies, replicative lifespan (RLS) and chronological lifespan (CLS) [4]. All eukaryotic cells undergo a finite number of cell divisions, before irreversibly exiting mitosis. RLS measures the number of daughter cells produced by a mother cell before senescence and is a model for studying aging of mitotically active cells. CLS determines the time that cells survive in a non-dividing state after depletion of nutrients in stationary phase and is a model for the study of post-mitotic cells. The fact that chronologically old yeast also have a shorter RLS [5][6] indicates that RLS and CLS share core mechanisms.
INTRODUCTION TO THE YEAST INTEGRATED STRESS RESPONSE
Strikingly, most interventions that extend lifespan are, or induce, limited amounts of stress that have a beneficial effect via the phenomenon of hormesis, because these stresses would be toxic or lethal at higher doses. These lifespan-extending hormetic stresses induce a protective cellular stress response. This conserved stress response in yeast was termed the General Amino Acid Control (GAAC) because it was initially identified as a response to amino acid depletion that upregulates the genes required for amino acid synthesis [7]. However, we now know that this same yeast stress response pathway is also activated by starvation for purines [8], glucose limitation/depletion [9], ER stress [10], growth on non-fermentable carbon sources like ethanol [9], high salt [11], treatment with alkylating agents [12], and TOR inhibition [13][14]. As such, here we refer to this yeast stress response pathway by the name used in multicelluar eukaryotes, the Integrated Stress Response (ISR) [15]. As discussed below, multiple different lifespan-extending regimens require the ISR for lifespan extension. As the ISR has been reviewed extensively recently [16], here we focus on the contribution of the yeast ISR to lifespan extension. Other stress responses, such as the retrograde response, have also been implicated in the extension of CLS and RLS of yeast, and have been extensively reviewed [17][18], so will not be discussed here.
–
During the ISR in yeast, a variety of stressful conditions activate the kinase Gcn2, which phosphorylates eIF2α, the central regulation node of the ISR. Phosphorylation of eIF2α promotes a stress-resistant state by global attenuation of protein synthesis, which reduces the load of unfolded proteins on chaperone networks and diverts amino acids from energetically costly protein synthesis to other metabolic pathways. Although the mammalian ISR has three other eIF2α kinases in addition to Gcn2, the stress response pathways are otherwise very similar in yeast and mammals [16]. Specifically, eIF2α phosphorylation downregulates formation of the eIF2 complex, resulting in reduced formation of the ternary complex containing eIF2, GTP and the initiator Methionyl-tRNA (Met-tRNAiMet). This in turn reduces delivery of Met-tRNAiMet to the 40S ribosome, hampering initiation of global cap-dependent translation [19] (Fig. 1). In addition to these general effects on protein and amino acid metabolism, eIF2α phosphorylation also serves to activate a gene expression program that depends on translational upregulation of Gcn4. The mechanism for the paradoxical synthesis of the ISR effector protein Gcn4 in the face of global translational downregulation has been revealed by the elegant work of the Hinnebusch lab [19]. Under normal conditions, translation of the short upstream open reading frames (uORFs) within the GCN4 mRNA leader occurs, followed by the ribosomes dissociating from the GCN4 mRNA, precluding initiation at the GCN4 start codon (Fig. 1). However, under conditions where there are limiting amounts of 40S ribosome associated with Met-tRNAiMet, the ribosomes scan through the inhibitory uORFs within the GCN4 leader and initiate translation at the GCN4 start codon. The mammalian counterpart of Gcn4, ATF4, is induced by an analogous mode of translational control [20].
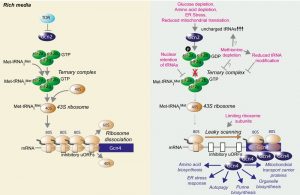 | FIGURE 1: Overview of the yeast integrated stress response and how it is influenced by lifespan extending interventions. During growth in nutrient rich conditions, the ternary complex is formed from eIF2, methionine bound to tRNAiMet, and GTP, which then delivers Met-tRNAiMet to the 40S ribosome to make the 43S complex, which is a key rate-limiting step in translational initiation. Under rich growth conditions, efficient translation of the inhibitory upstream ORFs (uORFs) of the GCN4 mRNA causes dissociation of the ribosome, preventing it scanning along the mRNA to the protein coding ORF (blue). The regimens indicated in pink, which all extend RLS, either result in increased abundance of uncharged tRNAs that activate Gcn2 to phosphorylate eIF2α to prevent formation of functional eIF2-GTP, or they activate Gcn4 in a Gcn2-independent manner, in conditions that reduce formation of the ternary complex directly (reduced amounts of processed tRNAs in the cytoplasm, reduced amino acids) or via reduced amounts of ribosomal subunits. In either case, the end result is inefficient 80S ribosome formation, which promotes leaky scanning along the mRNA to the protein coding ORF of GCN4. The resulting Gcn4 protein induces transcription of genes required for the indicated stress response pathways. |
–
Gcn4 can also be synthesized in the absence of Gcn2 activation, by conditions that directly reduce global cap-dependent translation (Fig. 1). For example, Gcn4 is translated in yeast strains lacking Gcn2 upon rapid depletion of amino acids [21], which presumably limits the formation of the ternary complex. Mutants that limit tRNA processing or tRNA nuclear export, such as los1∆, also activate Gcn4 in the absence of Gcn2, presumably because there is insufficient amount of ternary complex formation [22][23]. Similarly, reduced abundance of large ribosomal subunits could also trigger Gcn4 translation without the need for Gcn2 [24] as there would be less 60S ribosome to associate with the 40S-Met-tRNAiMet complex. Notably, amino acid depletion is a clear mechanism to extend both RLS and CLS, while reduced abundance of ribosomal subunits or deletion of the LOS1 gene required for tRNA export extend RLS in a manner dependent on Gcn4 but not Gcn2 [22][23][24].
–
The Gcn4 transcriptional activator upregulates genes that allow adaptation to cellular stress [16]. These include genes that mediate amino acid biosynthesis, purine biosynthesis, organelle biosynthesis, ER stress response, mitochondrial carrier proteins and autophagy [16] (Fig. 1). Although multiple stresses converge on eIF2α phosphorylation, the cellular outcome is not always the same. The effect of ISR activation depends on the nature of the stress, its duration, and the amount of stress, which in turn affects the extent of eIF2α phosphorylation and Gcn4 synthesis. A short-lived ISR is usually an adaptive response aimed at restoring homeostasis, while prolonged ISR activation can signal for cell death.
–
We are still at an early stage in trying to understand which of the downstream stress response pathways induced by Gcn4 are key to the lifespan extension mediated by the ISR (Fig. 1). Although not conclusive, some clues as to the relative importance of the different ISR-induced stress response pathways to lifespan can be provided by the phenotypes resulting from deletion of genes activated by the ISR. For example, it is unlikely that purine biosynthesis is key for lifespan extension, given that deletion of purine biosynthesis genes extends CLS [25]. Similarly, RLS is extended by deletion of various genes involved in amino acid synthesis [23] while deletion of multiple unfolded protein response (UPR) target genes extends RLS [26]. Furthermore, mitochondrial carrier proteins are degraded via a novel selective autophagy pathway in replicatively-aged yeast [27], warranting investigation into their role in lifespan determination. As such, the beneficial effects of inactivation of key genes in these ISR-target pathways are not consistent with their transcriptional induction extending lifespan. In further support of such an idea, the UPR is not even required for the lifespan extension that occurs in response to ER stress [28], as discussed in more detail below. By default, that leaves autophagy as the most promising target of the ISR for lifespan extension. By coincidence, autophagy has become of particular interest to the aging field [29][30].
–
Autophagy is a conserved catabolic housekeeping mechanism whereby bulk cytoplasm, including proteins and organelles, are sequestered into membrane-bound vesicles (autophagosomes) which subsequently fuse with vacuoles where their content is degraded by vacuolar proteases [31]. The identification of the autophagy genes and the mechanism of autophagy, like many key processes, occurred in yeast, in this case leading to the 2016 Nobel prizes in Physiology or Medicine [32]. Many of the genes and mechanisms are highly conserved in metazoans, including humans, with abnormalities in higher eukaryote autophagy resulting in the development of aging and age-related diseases, as previously reviewed [29][33]. Elsewhere, we have recently reviewed the contributions of autophagy to yeast lifespan extension [34]. Here, we synthesize the recent studies that link the ISR to lifespan extension in yeast, to support our proposal that the mechanism whereby stress extends lifespan is via induction of the ISR and downstream stress response pathways, including autophagy. By extension, we suggest that therapeutics that specifically induce the ISR and/or autophagy should be developed to promote human healthspan and extend lifespan.
THE ROLE OF THE ISR IN YEAST LIFESPAN EXTENSION
Nutrient depletion extends RLS and activates the ISR
The yeast RLS assay utilizes the asymmetric division of mother and daughter yeast cells to track the replicative capacity of individual cells, and has been used to identify various types of factors that contribute to replicative senescence. Comparison of daughter cells and old mother cells has revealed that many factors selectively accumulate in the mother cells, and these so-called senescence factors have been suggested to drive the aging process. These senescence factors include extra-ribosomal DNA circles [35], damaged nuclear pore complexes [36], cytosolic aggregates, oxidized proteins [37], and dysfunctional mitochondria [38]. In essence, mechanisms exist to exclude the senescence factors out of the daughter cells, to enable them to be rejuvenated while at the same time, causing the mother cells to age. In theory, any condition that results in the activation of autophagy to remove accumulating senescence factors, including damaged or aggregated proteins and damaged organelles and to recycle cellular components, would be a logical approach to extend the RLS of mother cells. Where examined, this is transpiring to be the case.
–
There is currently no evidence to suggest that either Gcn4 or autophagy contribute to longevity under nutrient-rich growth conditions. This is consistent with the fact that deletion of most ATG genes or GCN4 has minimal effects on RLS [23][28]. However, many of the interventions that extend yeast RLS also induce the ISR and/or autophagy. We propose that these RLS extending regimens extend lifespan via inducing the ISR and/or autophagy, as discussed below. Notably, many of the therapeutic, genetic or environmental interventions that extend yeast RLS are either a decrease of nutrients per se or they decrease the activity of the nutrient signaling pathways. Accordingly, inactivation of either of the two major nutrient signaling pathways extends yeast RLS: the target of rapamycin (TOR) / serine-threonine kinase Sch9 pathway [39] and the RAS / cAMP-dependent protein kinase A (PKA) pathway [40]. Exactly how these pathways regulate RLS is unclear, but in general, when nutrients are abundant, these nutrient sensing pathways inhibit the ISR and autophagy (Fig. 1). However, when nutrients are limiting, the nutrient sensing pathways are inactivated, their inhibitory influence on autophagy and the ISR is released, and yeast RLS is extended. So far, the relationship between RLS extension and autophagy induction upon inactivation of the nutrient sensing pathways is purely correlation. It has not yet been tested whether the induction of autophagy upon inhibition of the TOR/Sch9 or RAS/cAMP pathways is required for RLS extension, or whether there is any involvement of the ISR in their ability to extend RLS.
–
The role of the yeast ISR in autophagy
Gcn2 and Gcn4 were initially described as being required for the induction of autophagy upon nitrogen starvation. Specifically, deletion of GCN2 or GCN4 blocked nitrogen starvation-induced macroautophagy, as determined by electron micrography of large autophagosomes [41]. Closer examination in a later study uncovered that in response to nitrogen depletion, deletion of GCN2 or GCN4 only partially diminished autophagic flux, but formed multiple small autophagosomes, rather than large autophagosomes [42]. This phenotype was due to failure to transcriptionally upregulate Atg8 levels in the absence of Gcn2/Gcn4. In agreement with the fact that Gcn4 is induced at the translational level, the signaling mechanism that induces the initiation of macroautophagy occurs independent of new protein synthesis, while protein synthesis is required for the formation of large autophagosomes [43]. Autophagosome size depends on the transcriptional upregulation of Atg8 [44], which requires Gcn4 [42]. As such, Gcn2/Gcn4 are required for enlargement of autophagosomes, but not for autophagy per se upon nitrogen starvation. Gcn4 is also required for ATG1 and ATG32 transcription upon nitrogen starvation [45], where Atg1 is required for the initiation of autophagy and Atg32 is specifically required for mitophagy, the form of autophagy that selectively degrades mitochondria. Furthermore, many ATG genes have Gcn4 binding sites in their promoters [45]. Although nitrogen starvation is the conventional method to induce autophagy, its relevance to RLS is negligible, given that nitrogen starvation induces cell cycle arrest.
–
Amino acid depletion is another common mechanism used to induce autophagy. Notably, amino acid depletion is not only a mechanism to extend yeast RLS [46][47], but Gcn2/Gcn4 are also essential for macroautophagy upon amino acid depletion [42]. Amino acid starvation-induced autophagy responded to the levels of intracellular, not extracellular, amino acids, and Gcn2/Gcn4 were specifically found to be required for autophagic trafficking in response to amino acid starvation [42]. The reason why amino acid depletion-triggered autophagy, but not nitrogen starvation-triggered autophagy, absolutely requires Gcn2/Gcn4 is unclear as yet.
–
The requirement for the Gcn4 activator in many regimens of RLS extension
Although Gcn2 is conventionally activated by tRNAs that are not bound to amino acids (uncharged tRNAs), Gcn4 synthesis is induced under many conditions besides amino acid depletion. These include starvation for purines [8], glucose limitation [9], ER stress [10], growth on non-fermentable carbon sources like ethanol [9], high salt [11], treatment with alkylating agents [12] and TOR inhibition [13][14]. All these conditions that activate Gcn4 depend on the Gcn2 kinase and many extend yeast RLS. The mechanism whereby glucose limitation activates Gcn2 appears to be partially due to the depletion of cytoplasmic pools of amino acids that occurs upon glucose depletion. Accordingly, supplementation with amino acids reduces the Gcn4 synthesis that results from glucose limitation [9]. However, the effect is only partial, suggesting that mechanisms of Gcn2 activation that do not rely on uncharged tRNAs may also be at play.
–
The TOR pathway and the ISR pathway appear to be interlinked, although this is somewhat controversial. One report showed that the activation of autophagy that is triggered by rapamycin-mediated inhibition of TOR was not affected by deletion of GCN2 [41]. By contrast, RLS extension observed upon TOR1 deletion was mediated at least partially by Gcn4 [24], because it was reduced upon GCN4 deletion. The reason for the discrepancy between the two studies could reflect that, although Gcn2 is activated upon TOR inhibition, Gcn2 activation may not be essential for Gcn4 translation upon TOR inhibition. The reasoning for this is that there could be reduced translation of the uORFs in the GCN4 leader upon TOR inhibition or TOR1 deletion as a consequence of loss of TOR-mediated phosphorylation of the eukaryotic initiation factor eIF4E binding protein 4E-BP1 and accompanying down regulation of global protein synthesis. Alternatively, this could be due to potential changes in the small ribosomal subunit caused by decreased phosphorylation of ribosomal protein S6 (RpS6) by Sch9/S6K, which might interfere with 40S structure, changing its activity and increasing the leakiness of translation on the GCN4 mRNA. As discussed above, Gcn4 can be activated in the absence of Gcn2, in conditions where protein synthesis is reduced (Fig. 1). There is also feedback between the two pathways at least in one direction because inhibition of TOR signaling induces activation of Gcn2 [13][14] (Fig. 1). As such, it would appear that inhibition of the TOR pathway stimulates autophagy by not only releasing the inhibitory phosphorylation on Atg13 [48][49], but also by increasing Gcn4 synthesis to transcriptionally upregulate ATG genes (Fig. 1). Whether Gcn4 synthesis per se is sufficient to extend RLS or induce autophagy remains untested.
–
Disruptions of the ER-Golgi apparatus can extend RLS by inducing autophagy. In fact, this provides the most compelling evidence to date for a role for autophagy in RLS extension. An innovative genetic screen for deletion mutants that maintain the transcriptional silencing that is usually lost in old cells identified novel gene deletions that extend RLS [28]. One of these, RER1 encodes a receptor that maintains ER compartmentalization, and upon deletion extended RLS by 64% [28]. Deletion of RER1 resulted in ER stress, as it activated the UPR and autophagy, which are likely to mitigate an increase in ER load. In agreement with lifespan extension being caused by ER stress, the drug tunicamycin, which also activates ER stress, extends RLS. The autophagy induced by RER1 deletion was required for RLS extension, because deletion of genes encoding Atg8, or Atg5, the latter of which mediates the Atg8 lipidation required for expansion of autophagosomes, abolished the otherwise extended RLS in rer1∆ yeast. However, the UPR induced by ER stress was not required for RLS extension, because deletion of the IRE1 gene, encoding the principle sensor of the UPR, did not block RLS extension. The screen also recovered MNT3, encoding a Golgi glycosylase as being long-lived upon its deletion. Deletion of MNT3 led to high basal levels of UPR, elevated autophagy and extended mitotic lifespan [28]. As such, induction of autophagy and the accompanying increase in RLS appears to be a broad adaptive response evoked by impaired protein modification or trafficking across the ER-Golgi network. Given that ER stress is known to induce Gcn4 [10], it is likely that the lifespan-extending autophagy that is induced by ER stress is dependent on Gcn4.
–
Polyamines, such as spermidine, play a role in longevity. Polyamine levels, including spermidine, decrease during aging. Notably, addition of spermidine extends yeast RLS when given to old cells, but not young cells, isolated by elutriation [50]. Spermidine treatment increases autophagic flux, which may be related to the fact that spermidine leads to transcriptional upregulation of ATG7, ATG11, and ATG15, potentially through its influence on histone acetylation [50]. In agreement with the observed induction of autophagic flux, spermidine treatment promotes the removal of oxidized prions from yeast [51]. It is not currently known whether the extension of RLS by spermidine is mediated via inducing autophagy, although this appears to be the case with CLS [50]. Interestingly, the ability of spermidine to induce transcription of other stress-induced genes, such as the gene encoding the drug:H+ antiporter Qdr3, is dependent on the transcription factor Gcn4 [52]. It is tempting to speculate that spermidine-mediated yeast RLS extension also requires Gcn4, and potentially, Gcn4-dependent autophagy.
–
Gcn4 is required for RLS extension that occurs upon nuclear retention of tRNAs. Deletion of the gene encoding the tRNA exporter Los1 was found to extend RLS in a systematic analysis of 4,698 single gene deletions for the effect on the yeast RLS [23]. The lifespan extension upon LOS1 deletion was shown to be in the same pathway as lifespan extension by dietary restriction (DR) via epistasis analyses [23], suggesting that nuclear retention of tRNAs may be one of the mechanisms by which DR extends RLS. Deletion of LOS1 leads to the activation of Gcn4, reflected in the upregulation of many of its transcriptional targets, and seemingly in the absence of Gcn2 activation, because no global decrease in mRNA translation occurs [23]. The limited availability of tRNAs in the cytoplasm would presumably reduce formation of Met-tRNAiMet which would promote scanning through the Gcn4 upstream ORFs and Gcn4 synthesis (Fig. 1) without the need for Gcn2 activation. It will be interesting to discern if RLS extension upon LOS1 deletion is dependent on autophagy or other pathways induced by Gcn4.
–
Gcn4 is required for RLS extension that results from deletion of specific mitochondrial components. Yeast lacking the gene encoding the mitochondrial AAA protease Afg3 are long-lived [53]. The Afg3 protease has multiple functions, including helping to mature a mitochondrial ribosomal protein. This is reminiscent of RLS extension observed in yeast lacking the mitochondrial ribosomal protein Afo1 [54]. Furthermore, downregulation of mitochondrial protein synthesis by erythromycin extends RLS [55]. Taken together, these studies suggest that reduced mitochondrial translation can extend RLS; and in the case of Afg3, RLS extension was dependent on Gcn4, and AFG3 deletion reduced cellular mRNA translation [53]. It is possible that reduced mitochondrial translation activates the ISR, which results in reduced mRNA translation and extended RLS.
–
Requirement for the ISR in extension of CLS
Investigations into the involvement of Gcn2/Gcn4 in yeast CLS extension to date are limited, but are consistent with a role for the ISR in CLS extension. The Chinese herbal compound cryptotanshione extends CLS, but only when there are ample essential amino acids, and this is seemingly through the amino acid sensing pathways [56]. Specifically, cryptotanshione inactivates the Tor/Sch9 pathway, leading to elevated Gcn2 activity via release from Tor/Sch9-dependent inhibition (Fig. 1). Demonstrating the importance of these pathways for CLS extension by cryptotanshione, deletion of TOR1, SCH9 or GCN2 blocked cryptotanshione from extending CLS [56]. Methionine restriction extends organismal lifespan across many different species of eukaryotes [57]. Limiting amounts of methionine in synthetic defined media extends yeast CLS in a manner dependent on GCN2, but independent of TOR1 [58]. The mechanism at play appears to be due to increased levels of uncharged tRNAs that result from methionine depletion [59], activating Gcn2 and the ISR (Fig. 1). Addition, not depletion, of other specific amino acids to minimal media has been shown to extend CLS, and deletion of GCN4 blocks this CLS extension [60]. As such, there are clear examples of the ISR being required for CLS extension. Whether these mechanisms are through autophagy or other downstream responses of the ISR is unknown at present.
SUMMARY AND REMAINING QUESTIONS
Yeast aging models have been indispensable for elucidating mechanisms of cellular aging, especially with regards to nutrient signaling pathways. During an organism’s lifetime, aging cells undergo a multitude of changes and accumulate macromolecular damage. Decrease in high-nutrient PKA/Sch9/TOR signaling pathways leads to a slowing of the aging processes, likely due to the increased stress resistance and the regulation of proteostatic mechanisms. In this review we provide an overview of emerging evidence indicating the importance of the ISR as a parallel, yet opposing, nutrient-signaling mechanism to TOR signaling. Also, we highlight findings suggesting a fundamental role of the ISR as a target effector of nutrient signaling on yeast lifespan.
–
Although growing evidence suggests that the ISR is required for lifespan extension, we do not yet know if the ISR is sufficient for lifespan extension. Furthermore, is Gcn4 activation sufficient to extend yeast RLS or CLS? If this were the case, it would suggest that the general inhibition of cap-dependent translation is not required for yeast lifespan extension: rather it is the resulting upregulation of stress response gene expression that is key. As such, increasing Gcn4/ATF4 protein levels may be a promising therapeutic intervention to extend lifespan and healthspan without the detrimental effects of reduced global protein synthesis and immunosuppression that occur in humans upon TOR inhibition by compounds such as rapamycin. Notably, it is not yet clear whether the autophagy that is induced by lifespan extension regimens is dependent on Gcn4, or other autophagy-inducing pathways. Thus, therapeutics to directly activate autophagy could be another promising approach to extend lifespan and healthspan.
–
Gcn4 induces transcription of limiting components of multiple different stress response pathways (Fig. 1). This raises the question: is autophagy the only downstream process activated by Gcn4 that is required for lifespan extension? This could be answered by testing whether induction of autophagy is sufficient to extend yeast lifespan. This is clearly the case in multicellular eukaryotes because overexpression of Atg5 in mice, which increases autophagic flux, is sufficient to extend lifespan [61]. There are many different types of autophagy, and a better understanding of the aging process would be gained by knowledge of which specific autophagy pathways need to be activated to extend yeast lifespan.
–
The phenomenal pace of research and discoveries being made using the yeast model organism indicate that it will continue to be a central model for aging studies for the foreseeable future. Critically, since many of the components of longevity-regulating mechanisms are highly conserved from yeast to higher eukaryotes, the discovery of pharmaceutical and physiological regulators of these processes in yeast will likely have biological significance to human health and aging.
References
- K.J. Bitterman, O. Medvedik, and D.A. Sinclair, "Longevity regulation in Saccharomyces cerevisiae: linking metabolism, genome stability, and heterochromatin.", Microbiology and molecular biology reviews : MMBR, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12966141
- K. Steinkraus, M. Kaeberlein, and B. Kennedy, "Replicative Aging in Yeast: The Means to the End", Annual Review of Cell and Developmental Biology, vol. 24, pp. 29-54, 2008. http://dx.doi.org/10.1146/annurev.cellbio.23.090506.123509
- G. Janssens, and L. Veenhoff, "Evidence for the hallmarks of human aging in replicatively aging yeast", Microbial Cell, vol. 3, pp. 263-274, 2016. http://dx.doi.org/10.15698/mic2016.07.510
- V. Longo, G. Shadel, M. Kaeberlein, and B. Kennedy, "Replicative and Chronological Aging in Saccharomyces cerevisiae", Cell Metabolism, vol. 16, pp. 18-31, 2012. http://dx.doi.org/10.1016/j.cmet.2012.06.002
- C.R. Burtner, C.J. Murakami, B.K. Kennedy, and M. Kaeberlein, "A molecular mechanism of chronological aging in yeast", Cell Cycle, vol. 8, pp. 1256-1270, 2009. http://dx.doi.org/10.4161/cc.8.8.8287
- P. Fabrizio, L. Battistella, R. Vardavas, C. Gattazzo, L. Liou, A. Diaspro, J.W. Dossen, E.B. Gralla, and V.D. Longo, "Superoxide is a mediator of an altruistic aging program in Saccharomyces cerevisiae ", The Journal of Cell Biology, vol. 166, pp. 1055-1067, 2004. http://dx.doi.org/10.1083/jcb.200404002
- A.G. Hinnebusch, and G.R. Fink, "Positive regulation in the general amino acid control of Saccharomyces cerevisiae.", Proceedings of the National Academy of Sciences of the United States of America, 1983. http://www.ncbi.nlm.nih.gov/pubmed/6351059
- R.J. Rolfes, and A.G. Hinnebusch, "Translation of the yeast transcriptional activator GCN4 is stimulated by purine limitation: implications for activation of the protein kinase GCN2.", Molecular and cellular biology, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8336737
- R. Yang, S.A. Wek, and R.C. Wek, "Glucose limitation induces GCN4 translation by activation of Gcn2 protein kinase.", Molecular and cellular biology, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10733573
- O. Deloche, J. de la Cruz, D. Kressler, M. Doère, and P. Linder, "A membrane transport defect leads to a rapid attenuation of translation initiation in Saccharomyces cerevisiae.", Molecular cell, 2004. http://www.ncbi.nlm.nih.gov/pubmed/14967143
- A. Goossens, T.E. Dever, A. Pascual-Ahuir, and R. Serrano, "The Protein Kinase Gcn2p Mediates Sodium Toxicity in Yeast", Journal of Biological Chemistry, vol. 276, pp. 30753-30760, 2001. http://dx.doi.org/10.1074/jbc.M102960200
- K. Natarajan, M.R. Meyer, B.M. Jackson, D. Slade, C. Roberts, A.G. Hinnebusch, and M.J. Marton, "Transcriptional Profiling Shows that Gcn4p Is a Master Regulator of Gene Expression during Amino Acid Starvation in Yeast", Molecular and Cellular Biology, vol. 21, pp. 4347-4368, 2001. http://dx.doi.org/10.1128/MCB.21.13.4347-4368.2001
- V.A. Cherkasova, and A.G. Hinnebusch, "Translational control by TOR and TAP42 through dephosphorylation of eIF2α kinase GCN2", Genes & Development, vol. 17, pp. 859-872, 2003. http://dx.doi.org/10.1101/gad.1069003
- H. Kubota, T. Obata, K. Ota, T. Sasaki, and T. Ito, "Rapamycin-induced Translational Derepression of GCN4 mRNA Involves a Novel Mechanism for Activation of the eIF2α Kinase GCN2", Journal of Biological Chemistry, vol. 278, pp. 20457-20460, 2003. http://dx.doi.org/10.1074/jbc.C300133200
- H.P. Harding, Y. Zhang, H. Zeng, I. Novoa, P.D. Lu, M. Calfon, N. Sadri, C. Yun, B. Popko, R. Paules, D.F. Stojdl, J.C. Bell, T. Hettmann, J.M. Leiden, and D. Ron, "An integrated stress response regulates amino acid metabolism and resistance to oxidative stress.", Molecular cell, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12667446
- K. Pakos‐Zebrucka, I. Koryga, K. Mnich, M. Ljujic, A. Samali, and A.M. Gorman, "The integrated stress response", EMBO reports, vol. 17, pp. 1374-1395, 2016. http://dx.doi.org/10.15252/embr.201642195
- P.M. Quirós, A. Mottis, and J. Auwerx, "Mitonuclear communication in homeostasis and stress", Nature Reviews Molecular Cell Biology, vol. 17, pp. 213-226, 2016. http://dx.doi.org/10.1038/nrm.2016.23
- S.M. Jazwinski, "The Retrograde Response", Progress in Molecular Biology and Translational Science, pp. 133-154, 2014. http://dx.doi.org/10.1016/B978-0-12-394625-6.00005-2
- A.G. Hinnebusch, "Translational regulation of GCN4 and the general amino acid control of yeast.", Annual review of microbiology, 2005. http://www.ncbi.nlm.nih.gov/pubmed/16153175
- R.J. Jackson, C.U.T. Hellen, and T.V. Pestova, "The mechanism of eukaryotic translation initiation and principles of its regulation", Nature Reviews Molecular Cell Biology, vol. 11, pp. 113-127, 2010. http://dx.doi.org/10.1038/nrm2838
- D. Tzamarias, I. Roussou, and G. Thireos, "Coupling of GCN4 mRNA translational activation with decreased rates of polypeptide chain initiation.", Cell, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2661015
- H. Qiu, C. Hu, J. Anderson, G.R. Björk, S. Sarkar, A.K. Hopper, and A.G. Hinnebusch, "Defects in tRNA processing and nuclear export induce GCN4 translation independently of phosphorylation of the alpha subunit of eukaryotic translation initiation factor 2.", Molecular and cellular biology, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10713174
- M. McCormick, J. Delaney, M. Tsuchiya, S. Tsuchiyama, A. Shemorry, S. Sim, A. Chou, U. Ahmed, D. Carr, C. Murakami, J. Schleit, G. Sutphin, B. Wasko, C. Bennett, A. Wang, B. Olsen, R. Beyer, T. Bammler, D. Prunkard, S. Johnson, J. Pennypacker, E. An, A. Anies, A. Castanza, E. Choi, N. Dang, S. Enerio, M. Fletcher, L. Fox, S. Goswami, S. Higgins, M. Holmberg, D. Hu, J. Hui, M. Jelic, K. Jeong, E. Johnston, E. Kerr, J. Kim, D. Kim, K. Kirkland, S. Klum, S. Kotireddy, E. Liao, M. Lim, M. Lin, W. Lo, D. Lockshon, H. Miller, R. Moller, B. Muller, J. Oakes, D. Pak, Z. Peng, K. Pham, T. Pollard, P. Pradeep, D. Pruett, D. Rai, B. Robison, A. Rodriguez, B. Ros, M. Sage, M. Singh, E. Smith, K. Snead, A. Solanky, B. Spector, K. Steffen, B. Tchao, M. Ting, H. Vander Wende, D. Wang, K. Welton, E. Westman, R. Brem, X. Liu, Y. Suh, Z. Zhou, M. Kaeberlein, and B. Kennedy, "A Comprehensive Analysis of Replicative Lifespan in 4,698 Single-Gene Deletion Strains Uncovers Conserved Mechanisms of Aging", Cell Metabolism, vol. 22, pp. 895-906, 2015. http://dx.doi.org/10.1016/j.cmet.2015.09.008
- K.K. Steffen, V.L. MacKay, E.O. Kerr, M. Tsuchiya, D. Hu, L.A. Fox, N. Dang, E.D. Johnston, J.A. Oakes, B.N. Tchao, D.N. Pak, S. Fields, B.K. Kennedy, and M. Kaeberlein, "Yeast Life Span Extension by Depletion of 60S Ribosomal Subunits Is Mediated by Gcn4", Cell, vol. 133, pp. 292-302, 2008. http://dx.doi.org/10.1016/j.cell.2008.02.037
- M. Matecic, D.L. Smith, X. Pan, N. Maqani, S. Bekiranov, J.D. Boeke, and J.S. Smith, "A Microarray-Based Genetic Screen for Yeast Chronological Aging Factors", PLoS Genetics, vol. 6, pp. e1000921, 2010. http://dx.doi.org/10.1371/journal.pgen.1000921
- V.M. Labunskyy, M.V. Gerashchenko, J.R. Delaney, A. Kaya, B.K. Kennedy, M. Kaeberlein, and V.N. Gladyshev, "Lifespan Extension Conferred by Endoplasmic Reticulum Secretory Pathway Deficiency Requires Induction of the Unfolded Protein Response", PLoS Genetics, vol. 10, pp. e1004019, 2014. http://dx.doi.org/10.1371/journal.pgen.1004019
- A.L. Hughes, C.E. Hughes, K.A. Henderson, N. Yazvenko, and D.E. Gottschling, "Selective sorting and destruction of mitochondrial membrane proteins in aged yeast", eLife, vol. 5, 2016. http://dx.doi.org/10.7554/eLife.13943
- A. Ghavidel, K. Baxi, V. Ignatchenko, M. Prusinkiewicz, T.G. Arnason, T. Kislinger, C.E. Carvalho, and T.A.A. Harkness, "A Genome Scale Screen for Mutants with Delayed Exit from Mitosis: Ire1-Independent Induction of Autophagy Integrates ER Homeostasis into Mitotic Lifespan", PLOS Genetics, vol. 11, pp. e1005429, 2015. http://dx.doi.org/10.1371/journal.pgen.1005429
- S. Gelino, and M. Hansen, "Autophagy - An Emerging Anti-Aging Mechanism.", Journal of clinical & experimental pathology, 2012. http://www.ncbi.nlm.nih.gov/pubmed/23750326
- F. Madeo, A. Zimmermann, M.C. Maiuri, and G. Kroemer, "Essential role for autophagy in life span extension", Journal of Clinical Investigation, vol. 125, pp. 85-93, 2015. http://dx.doi.org/10.1172/JCI73946
- J. Kaur, and J. Debnath, "Autophagy at the crossroads of catabolism and anabolism", Nature Reviews Molecular Cell Biology, vol. 16, pp. 461-472, 2015. http://dx.doi.org/10.1038/nrm4024
- S.A. Tooze, and I. Dikic, "Autophagy Captures the Nobel Prize", Cell, vol. 167, pp. 1433-1435, 2016. http://dx.doi.org/10.1016/j.cell.2016.11.023
- R.T. Netea-Maier, T.S. Plantinga, F.L. van de Veerdonk, J.W. Smit, and M.G. Netea, "Modulation of inflammation by autophagy: Consequences for human disease", Autophagy, vol. 12, pp. 245-260, 2016. http://dx.doi.org/10.1080/15548627.2015.1071759
- J. Tyler, "The role of autophagy in the regulation of yeast lifespan", Ann N Y Acad Sci: in press, 2017.
- D.A. Sinclair, and L. Guarente, "Extrachromosomal rDNA circles--a cause of aging in yeast.", Cell, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9428525
- Z. Shcheprova, S. Baldi, S.B. Frei, G. Gonnet, and Y. Barral, "A mechanism for asymmetric segregation of age during yeast budding", Nature, vol. 454, pp. 728-734, 2008. http://dx.doi.org/10.1038/nature07212
- H. Aguilaniu, L. Gustafsson, M. Rigoulet, and T. Nyström, "Asymmetric Inheritance of Oxidatively Damaged Proteins During Cytokinesis", Science, vol. 299, pp. 1751-1753, 2003. http://dx.doi.org/10.1126/science.1080418
- S. JAZWINSKI, "Yeast replicative life span – the mitochondrial connection", FEMS Yeast Research, vol. 5, pp. 119-125, 2004. http://dx.doi.org/10.1016/j.femsyr.2004.04.005
- M. Kaeberlein, R.W. Powers, K.K. Steffen, E.A. Westman, D. Hu, N. Dang, E.O. Kerr, K.T. Kirkland, S. Fields, and B.K. Kennedy, "Regulation of Yeast Replicative Life Span by TOR and Sch9 in Response to Nutrients", Science, vol. 310, pp. 1193-1196, 2005. http://dx.doi.org/10.1126/science.1115535
- S.J. Lin, P.A. Defossez, and L. Guarente, "Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae.", Science (New York, N.Y.), 2000. http://www.ncbi.nlm.nih.gov/pubmed/11000115
- Z. Tallóczy, W. Jiang, H.W. Virgin, D.A. Leib, D. Scheuner, R.J. Kaufman, E. Eskelinen, and B. Levine, "Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway", Proceedings of the National Academy of Sciences, vol. 99, pp. 190-195, 2001. http://dx.doi.org/10.1073/pnas.012485299
- N. Ecker, A. Mor, D. Journo, and H. Abeliovich, "Induction of autophagic flux by amino acid deprivation is distinct from nitrogen starvation-induced macroautophagy", Autophagy, vol. 6, pp. 879-890, 2010. http://dx.doi.org/10.4161/auto.6.7.12753
- H. Abeliovich, W.A. Dunn, J. Kim, and D.J. Klionsky, "Dissection of autophagosome biogenesis into distinct nucleation and expansion steps.", The Journal of cell biology, 2000. http://www.ncbi.nlm.nih.gov/pubmed/11086004
- H. Nakatogawa, Y. Ichimura, and Y. Ohsumi, "Atg8, a Ubiquitin-like Protein Required for Autophagosome Formation, Mediates Membrane Tethering and Hemifusion", Cell, vol. 130, pp. 165-178, 2007. http://dx.doi.org/10.1016/j.cell.2007.05.021
- A. Bernard, M. Jin, Z. Xu, and D.J. Klionsky, "A large-scale analysis of autophagy-related gene expression identifies new regulators of autophagy", Autophagy, vol. 11, pp. 2114-2122, 2015. http://dx.doi.org/10.1080/15548627.2015.1099796
- J.C. Jiang, E. Jaruga, M.V. Repnevskaya, and S.M. Jazwinski, "An intervention resembling caloric restriction prolongs life span and retards aging in yeast", The FASEB Journal, vol. 14, pp. 2135-2137, 2000. http://dx.doi.org/10.1096/fj.00-0242fje
- A. Kaya, B.C. Lee, and V.N. Gladyshev, "Regulation of Protein Function by Reversible Methionine Oxidation and the Role of Selenoprotein MsrB1", Antioxidants & Redox Signaling, vol. 23, pp. 814-822, 2015. http://dx.doi.org/10.1089/ars.2015.6385
- Y. Kamada, T. Funakoshi, T. Shintani, K. Nagano, M. Ohsumi, and Y. Ohsumi, "Tor-mediated induction of autophagy via an Apg1 protein kinase complex.", The Journal of cell biology, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10995454
- Y. Kamada, K. Yoshino, C. Kondo, T. Kawamata, N. Oshiro, K. Yonezawa, and Y. Ohsumi, "Tor Directly Controls the Atg1 Kinase Complex To Regulate Autophagy", Molecular and Cellular Biology, vol. 30, pp. 1049-1058, 2010. http://dx.doi.org/10.1128/MCB.01344-09
- T. Eisenberg, H. Knauer, A. Schauer, S. Büttner, C. Ruckenstuhl, D. Carmona-Gutierrez, J. Ring, S. Schroeder, C. Magnes, L. Antonacci, H. Fussi, L. Deszcz, R. Hartl, E. Schraml, A. Criollo, E. Megalou, D. Weiskopf, P. Laun, G. Heeren, M. Breitenbach, B. Grubeck-Loebenstein, E. Herker, B. Fahrenkrog, K. Fröhlich, F. Sinner, N. Tavernarakis, N. Minois, G. Kroemer, and F. Madeo, "Induction of autophagy by spermidine promotes longevity", Nature Cell Biology, vol. 11, pp. 1305-1314, 2009. http://dx.doi.org/10.1038/ncb1975
- S. Speldewinde, and C. Grant, "Spermidine cures yeast of prions", Microbial Cell, vol. 3, pp. 46-48, 2016. http://dx.doi.org/10.15698/mic2016.01.474
- M.C. Teixeira, T.R. Cabrito, Z.M. Hanif, R.C. Vargas, S. Tenreiro, and I. Sá-Correia, "Yeast response and tolerance to polyamine toxicity involving the drug : H+ antiporter Qdr3 and the transcription factors Yap1 and Gcn4", Microbiology, vol. 157, pp. 945-956, 2011. http://dx.doi.org/10.1099/mic.0.043661-0
-
J.R. Delaney, U. Ahmed, A. Chou, S. Sim, D. Carr, C.J. Murakami, J. Schleit, G.L. Sutphin, E.H. An, A. Castanza, M. Fletcher, S. Higgins, M. Jelic, S. Klum, B. Muller, Z.J. Peng, D. Rai, V. Ros, M. Singh, H.V. Wende, B.K. Kennedy, and M. Kaeberlein, "Stress profiling of longevity mutants identifies
Afg3 as a mitochondrial determinant of cytoplasmicmRNA translation and aging", Aging Cell, vol. 12, pp. 156-166, 2012. http://dx.doi.org/10.1111/acel.12032 - G. Heeren, M. Rinnerthaler, P. Laun, P. von Seyerl, S. Kössler, H. Klinger, S. Jarolim, B. Simon-Nobbe, M. Hager, C. Schüller, D. Carmona-Gutierrez, L. Breitenbach-Koller, C. Mück, P. Jansen-Dürr, A. Criollo, G. Kroemer, F. Madeo, and M. Breitenbach, "The mitochondrial ribosomal protein of the large subunit, Afo1p, determines cellular longevity through mitochondrial back-signaling via TOR1", Aging, vol. 1, pp. 622-636, 2009. http://dx.doi.org/10.18632/aging.100065
- M.A. Holbrook, and J.R. Menninger, "Erythromycin slows aging of Saccharomyces cerevisiae.", The journals of gerontology. Series A, Biological sciences and medical sciences, 2002. http://www.ncbi.nlm.nih.gov/pubmed/11773204
- Z. Wu, L. Song, S.Q. Liu, and D. Huang, "Tanshinones extend chronological lifespan in budding yeast Saccharomyces cerevisiae", Applied Microbiology and Biotechnology, vol. 98, pp. 8617-8628, 2014. http://dx.doi.org/10.1007/s00253-014-5890-5
- G.P. Ables, and J.E. Johnson, "Pleiotropic responses to methionine restriction", Experimental Gerontology, vol. 94, pp. 83-88, 2017. http://dx.doi.org/10.1016/j.exger.2017.01.012
- Z. Wu, L. Song, S.Q. Liu, and D. Huang, "Independent and Additive Effects of Glutamic Acid and Methionine on Yeast Longevity", PLoS ONE, vol. 8, pp. e79319, 2013. http://dx.doi.org/10.1371/journal.pone.0079319
- J.E. Johnson, and F.B. Johnson, "Methionine Restriction Activates the Retrograde Response and Confers Both Stress Tolerance and Lifespan Extension to Yeast, Mouse and Human Cells", PLoS ONE, vol. 9, pp. e97729, 2014. http://dx.doi.org/10.1371/journal.pone.0097729
- A.L. Alvers, L.K. Fishwick, M.S. Wood, D. Hu, H.S. Chung, W.A. Dunn Jr, and J.P. Aris, "Autophagy and amino acid homeostasis are required for chronological longevity in Saccharomyces cerevisiae", Aging Cell, vol. 8, pp. 353-369, 2009. http://dx.doi.org/10.1111/j.1474-9726.2009.00469.x
- J. Pyo, S. Yoo, H. Ahn, J. Nah, S. Hong, T. Kam, S. Jung, and Y. Jung, "Overexpression of Atg5 in mice activates autophagy and extends lifespan", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms3300
ACKNOWLEDGMENTS
This work was funded by NIH RO1 grants CA95641 and AG050660 to JT.
COPYRIGHT
© 2017

The integrated stress response in budding yeast lifespan extension by Postnikoff is licensed under a Creative Commons Attribution 4.0 International License.








