
Research Reports:
Microbial Cell, Vol. 3, No. 12, pp. 621 - 631; doi: 10.15698/mic2016.12.549
The transcription factors ADR1 or CAT8 are required for RTG pathway activation and evasion from yeast acetic acid-induced programmed cell death in raffinose
1 National Research Council of Italy, Institute of Biomembranes and Bioenergetics, Bari, Italy.
2 Department of Biological Sciences, University of New Orleans, New Orleans, LA, USA.
# L. L. and N. G. contributed equally to this work.
¶ Present address: Institute for Research on Cancer and Aging, CNRS-UMR 7284-INSERM U1081, University of Nice Sophia-Antipolis, Nice, France.
Keywords: yeast, programmed cell death, mitochondrial retrograde pathway, glucose, carbon catabolite repression, acetic acid.
Abbreviations:
AA-PCD - programmed cell death induced by acetic acid,
CCR -carbon catabolite repression,
GLU-WT - yeast cells grown in glucose,
PS - phosphatidylserine,
RAF-WT - yeast cells grown in raffinose,
ROS - reactive oxygen species,
TCA - tricarboxylic acid,
WT – wild-type.
Received originally: 20/02/2016 Received in revised form: 19/10/2016
Accepted: 26/10/2016
Published: 02/12/2016
Correspondence:
Sergio Giannattasio, s.giannattasio@ibbe.cnr.it
Conflict of interest statement:
The authors declare no conflict of interest.
Please cite this article as: Luna Laera, Nicoletta Guaragnella, Maša Ždralević, Domenico Marzulli, Zhengchang Liu and Sergio Giannattasio (2016). The transcription factors ADR1 or CAT8 are required for RTG pathway activation and evasion from yeast acetic acid-induced programmed cell death in raffinose. Microbial Cell 3(12): 621-631.
Abstract
Yeast Saccharomyces cerevisiae grown on glucose undergoes programmed cell death (PCD) induced by acetic acid (AA-PCD), but evades PCD when grown in raffinose. This is due to concomitant relief of carbon catabolite repression (CCR) and activation of mitochondrial retrograde signaling, a mitochondria-to-nucleus communication pathway causing up-regulation of various nuclear target genes, such as CIT2, encoding peroxisomal citrate synthase, dependent on the positive regulator RTG2 in response to mitochondrial dysfunction. CCR down-regulates genes mainly involved in mitochondrial respiratory metabolism. In this work, we investigated the relationships between the RTG and CCR pathways in the modulation of AA-PCD sensitivity under glucose repression or de-repression conditions. Yeast single and double mutants lacking RTG2 and/or certain factors regulating carbon source utilization, including MIG1, HXK2, ADR1, CAT8, and HAP4, have been analyzed for their survival and CIT2 expression after acetic acid treatment. ADR1 and CAT8 were identified as positive regulators of RTG-dependent gene transcription. ADR1 and CAT8 interact with RTG2 and with each other in inducing cell resistance to AA-PCD in raffinose and controlling the nature of cell death. In the absence of ADR1 and CAT8, AA-PCD evasion is acquired through activation of an alternative factor/pathway repressed by RTG2, suggesting that RTG2 may play a function in promoting necrotic cell death in repressing conditions when RTG pathway is inactive. Moreover, our data show that simultaneous mitochondrial retrograde pathway activation and SNF1-dependent relief of CCR have a key role in central carbon metabolism reprogramming which modulates the yeast acetic acid-stress response.
INTRODUCTION
Glucose is by far the preferred carbon source of the budding yeast Saccharomyces cerevisiae, because glucose metabolic regulation dictates the organism’s distinctive fermentative lifestyle—aerobic fermentation (the Crabtree effect) [1][2]. The phenomenon of glucose repression is a global regulatory mechanism causing inhibition of transcription of a large set of genes mainly involved in mitochondrial respiratory metabolism [3][4][5][6], which is known as the carbon catabolite repression (CCR) pathway. CCR is mediated, in part, by the crosstalk between two glucose signaling pathways: the RGT2/SNF3 axis responsible for glucose uptake [7][8][9]; and the SNF1/MIG1 axis that negatively regulates the genes involved in respiratory metabolism and the use of alternative sugars [3][10][11]. Since energy generation by fermentation is inefficient in terms of the ATP yield, yeast cells pump a large amount of glucose through glycolysis by enhancing its uptake, the first, rate-limiting step of glucose metabolism [12]. The CCR pathway interacts with other intracellular signaling pathways inducing a signaling network which senses the constantly fluctuating nutrient content of the environment, determining cell growth, stress resistance and metabolism [13].
–
Yeast cells grown in glucose (GLU-WT) undergo acetic acid-induced programmed cell death (AA-PCD) sharing many morphological and biochemical features with mammalian apoptosis, including DNA fragmentation, phosphatidylserine (PS) externalization and mitochondrial dysfunction (for ref see [14][15]). We have demonstrated that energy metabolism influences AA-PCD. Indeed, yeast cells grown in raffinose (RAF-WT) evade AA-PCD due to concomitant relief from CCR and activation of mitochondrial retrograde (RTG) signaling [16], a mitochondria-to-nucleus communication pathway causing up-regulation of a broad array of nuclear target genes in response to mitochondrial dysfunction dependent on RTG2 and MKS1, coding for positive and negative regulator of the pathway [17] (Fig. 1). The RTG pathway has been implicated in the intracellular signaling network linking mitochondrial function and cellular metabolism to several physiological processes such as ageing [18], PCD [16], autophagy [19] and ceramide metabolism [20].
–
There are two groups of RTG-target genes. The first group includes CIT2, encoding peroxisomal citrate synthase, and DLD3, encoding D-lactate dehydrogenase, whose transcription is strictly dependent on the heterodimeric transcription factor Rtg1/3 and shows a robust retrograde response [21]. The second group includes a number of tricarboxylic acid (TCA) cycle genes, which do not show an obvious retrograde response, their transcriptional control switching from the Hap2/3/4/5 complex to Rtg1/3 in response to a reduction or loss of respiratory function [22]. Hap2/3/4/5 complex is a transcription factor that is subject to glucose repression together with Adr1 and Cat8 [23][24] (Fig. 1). Hap4 regulates the expression of genes involved in mitochondrial respiratory metabolism including those encoding the TCA cycle enzymes and components of the respiratory chain complex genes [25][26][27]. Adr1 regulates genes involved in peroxisomal biogenesis, β-oxidation and utilization of non-fermentable carbon sources [28], whereas Cat8 regulates genes encoding gluconeogenic and glyoxylate cycle enzymes [29].
–
In this work, we used yeast to gain further insights into the relationships between the RTG and CCR pathways in the modulation of AA-PCD sensitivity under glucose repression or de-repression conditions, i.e. in glucose-grown (GLU-WT) cells, which undergo AA-PCD, or in raffinose-grown (RAF-WT) cells, which evade AA-PCD. To this aim we used yeast mutants lacking the RTG-signaling regulators RTG2, MKS1 and/or certain transcription factors regulating carbon source utilization.
RESULTS
Genetic inactivation of CCR by MIG1 or HXK2 deletion does not affect sensitivity of yeast to AA-PCD
Genes involved in CCR are of two types: genes such as HXK2 and MIG1, which are active in glucose-grown cells and repress certain transcription factors such as Hap4 and Cat8; genes required for de-repression, such as SNF1 and its downstream-activated transcription factors, Adr1 and Cat8, which are active in cells grown on alternative carbon sources, like raffinose (Fig. 1). In order to investigate the relations between glucose repression and the determination of cell fate, we first analyzed the effect of MIG1 or HXK2 deletions on AA-PCD sensitivity under glucose repression conditions. MIG1 encodes a DNA-binding zinc-finger protein that forms a part of a central transcriptional repressor complex, exerting its function on many genes, including those encoding for respiratory, gluconeogenic and alternative carbon source utilization proteins, whereas HXK2 encodes for hexokinase 2, the principal glucose phosphorylation enzyme and a putative glucose sensor [4]. Δmig1 and Δhxk2 mutant cells were treated with 80 mM acetic acid in a medium containing 2% glucose and cell survival was analyzed over time and compared to that of wild-type (WT) cells (Fig. 2A). Both Δmig1 and Δhxk2 mutant cells progressively lost their viability within 200 min, when more than 90% of the cells were unviable. Comparison with GLU-WT cells revealed a higher percentage of survival at 60 min (about 80% for Δmig1 and Δhxk2 mutants versus ~45% for WT), indicating a transient delay in cell death in these two mutants in response to acetic acid treatment. DNA fragmentation was analyzed in these cells to evaluate the nature of cell death. At 150 min, about 90% of Δmig1 or Δhxk2 cells were positive in the TUNEL assay (Fig. 2B), similar to TUNEL-positive GLU-WT cells (80%). In the absence of acetic-acid treatment, less than 5% of all three strains analyzed were TUNEL-positive. These data show that perturbations in glucose repression by deleting MIG1 or HXK2 did not affect final AA-PCD in glucose-grown cells. Then, we analyzed Δmig1Δmks1 and Δhxk2Δmks1 cells to study the effect of simultaneous constitutive activation of the RTG pathway and CCR inactivation on AA-PCD in glucose. We found that Δmig1Δmks1 cells completely lost viability like WT and single knock-out cells, whereas Δhxk2Δmks1 showed 20% survival after 200 min acetic acid treatment (Fig. 1S).
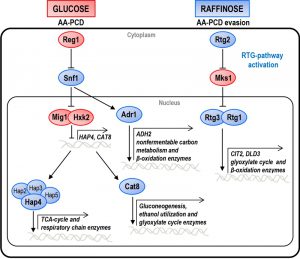 | FIGURE 1: CCR and RTG pathway network. Schematic overview of the CCR and RTG pathways in yeast cells growing either in glucose, in which cells undergo AA-PCD, or in raffinose, in which cells evade AA-PCD. For details see text. The factors regulating either pathway are indicated. Blue and red shapes indicate proteins active in raffinose or in glucose, respectively. Major genes whose transcription is controlled by each pathway are indicated. |
ADR1 and CAT8 cooperate with RTG2 to induce evasion of yeast AA-PCD in raffinose
In contrast to glucose-grown cells, raffinose-grown cells, in which mitochondrial respiration is de-repressed, evade AA-PCD [16]. HAP4, ADR1 and CAT8 encode transcriptional activators that control expression of glucose-repressible genes [30]. They are directly or indirectly activated by SNF1, the yeast homologue of AMP-activated protein kinase (AMPK), only under glucose depletion or with alternative carbon sources, when metabolism is shifted to aerobic respiration. Since SNF1 is an essential gene for cell growth in raffinose in W303-1B background strains, to investigate the role of glucose-repressible transcriptional factors in evasion of yeast AA-PCD under glucose de-repression conditions, viability of AA-treated Δhap4, Δadr1 or Δcat8 mutant cells was analyzed in raffinose as a function of time together with GLU-WT and RAF-WT cells, which undergo and evade AA-PCD, respectively, as controls (Fig. 2C). Δrtg2 mutant cells, which undergo AA-PCD due to inactivation of the RTG pathway [16], were also analyzed. Δadr1 and Δcat8 mutant cells progressively lost their viability reaching about 35% after 200 min, whereas, as expected, GLU-WT cells completely lost their viability, RAF-WT remained 100% viable and Δrtg2 cell viability decreased to about 50% at the same time point (Fig. 2C). Δhap4 cells were as viable as RAF-WT after 200 min, consistent with our published results [16]. Accordingly, the percentage of cells showing DNA fragmentation, as judged by TUNEL assay, was inversely proportional to cell viability at 150 min for all cell types analyzed (RAF-WT, 20%; Δadr1, 30%; Δcat8, 25%; GLU-WT, 80%) except in the case of Δrtg2 (47%) (Fig. 2D). Thus, RTG2, CAT8 and ADR1 are all important for cells to evade AA-PCD in raffinose as the carbon source.
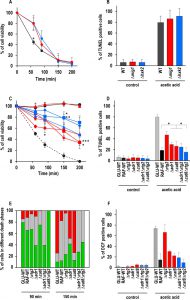 | FIGURE 2: Effect of genetic inactivation of both RTG and CCR pathways on AA-PCD in either glucose- or raffinose-grown cells. (A) Wild-type (WT, black), Δmig1 (red) and Δhxk2 (blue) mutant cells were treated with 80 mM acetic acid in growth medium with glucose as carbon source. Cell viability was analyzed by measuring colony-forming units (cfu) at indicated times. Cell survival based on the cfu was set at 100% at 0 min. The means of five independent experiments with standard deviations are reported. Anova-Bonferroni test: statistically different with (*) p < 0.001 when comparing WT with Δmig1 or Δhxk2 mutant cells. (B) DNA fragmentation in cells grown in glucose was detected by the TUNEL assay using confocal microscopy analysis. Percentage of TUNEL-positive cells is reported at 150 min. At least 400 cells were analyzed in three samples from each of three independent experiments. WT, grey bars; Δmig1, red bars; Δhxk2, blue bars. (C) GLU-WT (●, dashed line) and RAF-WT (●, black line), Δrtg2 (●, red line), Δadr1 (■, red line), Δcat8 (♦, red line), Δhap4 (▲, red line), Δadr1Δrtg2 (■, blue line), Δcat8Δrtg2 (♦, blue line) and Δhap4Δrtg2 (▲, blue line) cells were treated with acetic acid either in glucose or in raffinose as carbon source. Cell viability was analyzed at indicated times by measuring colony-forming units (cfu). Cell survival (100%) corresponds to the cfu at time zero. The means of five independent experiments with standard deviations are reported. Anova-Bonferroni test: statistically different with (*) p < 0.01 when comparing Δrtg2 versus Δcat8Δrtg2 at 90 min, Δadr1 with Δadr1Δrtg2 at 150 min or both Δadr1 and Δcat8 with Δcat8Δrtg2 at 200 min; (**) p < 0.001 when comparing Δcat8 with Δcat8Δrtg2 at 150 min; (***) p < 0.0001 when comparing Δadr1 with Δadr1Δrtg2 at 200 min. (D) DNA fragmentation was detected by TUNEL assay using confocal microscopy analysis. Percentage of TUNEL-positive cells is reported at 150 min. At least 400 cells were analyzed in three samples from each of three independent experiments. Fisher’s exact test: statistically different with (*) p < 0.01 when comparing Δadr1, Δcat8 or Δadr1Δrtg2 versus WT or Δcat8. GLU-WT, grey bars; RAF-WT, black bars; single knock-out cells, red bars; double knock-out cells, blue bars. (E) In a typical experiment GLU-WT, RAF-WT and knock-out cells grown in raffinose as indicated, were treated with acetic acid (AA). The cells were collected at 90 and 150 min after AA treatment, co-stained with Annexin V-FITC/PI and analyzed by confocal microscopy to measure PS externalization on the cell surface. The bars indicate the percentage of stained cells at different stages of death: early apoptotic cells (Annexin V+/PI-, green); late apoptotic cells (Annexin V+/PI+, grey); necrotic cells (Annexin V-/PI+, red). At least 400 cells were counted for each sample at indicated time. The experiment was repeated twice, with virtual identical results. Fisher’s exact test: statistically different with p < 0.005 when comparing Annexin V+ Δadr1 or Δadr1Δrtg2 versus all other cell types at 90 min, when comparing PI+ Δadr1 versus Δcat8 at 150 min and when comparing Annexin V+ Δadr1 or Δcat8Δrtg2 versus all other cell types at 150 min. (F) GLU-WT, RAF-WT and knock-out cells grown in raffinose, as indicated, were incubated in the absence (control) or in the presence of acetic acid for 30 min, then collected and stained with H2DCF-DA. DCF-stained cells due to ROS accumulation were analyzed by confocal microscopy. Bars indicate the percentage of DCF-positive cells with SD, calculated by counting at least 400 cells in three independent experiments. GLU-WT, grey bars; RAF-WT, black bars; single knock-out cells, red bars; double knock-out cells, blue bars. |
In order to investigate potential interactions between RTG2 and CAT8 or ADR1 in determining yeast cell evasion of AA-PCD, we generated Δadr1Δrtg2 and Δcat8Δrtg2 double mutant strains and determined cell survival en route to AA-PCD. Δhap4Δrtg2 cells [16] were also analyzed. Surprisingly, cell viability measured during AA treatment was significantly higher for Δadr1Δrtg2 and Δcat8Δrtg2 double mutants compared to the respective single mutants, with ~79% and 70% viability at 150 and 200 min, respectively, for the double mutants versus about 50% to 34% for the single mutants (Fig. 2C). The same behaviour was shown by Δadr1Δrtg3 and Δcat8Δrtg3 cells lacking the Rtg2-dependent transcriptional complex Rtg1/3 (Fig. 1S). Δhap4Δrtg2 cells lost viability essentially as with the Δrtg2 single mutant. However, whereas the Δcat8Δrtg2 double mutant showed a small but significant reduction in DNA fragmentation as compared with the Δcat8 and Δrtg2 single mutants, the percentage of Δadr1Δrtg2 TUNEL-positive cells was similar to that of Δadr1 cells (Fig. 2D). As expected, in the absence of AA-treatment, less than 5% of the cells were TUNEL-positive.
–
Since double knock-out cells showed virtually the same percentage of DNA fragmentation as single knock-out cells, although with a higher viability, we looked more closely at the nature of the death process in WT and knock-out cells and measured PS externalization on the cell surface and reactive oxygen species (ROS) accumulation. We first analyzed WT and knock-out cells en route to AA-induced cell death by Annexin V/PI co-staining and confocal microscopy as a function of time. PI staining of cells indicates loss of plasma membrane integrity. By Annexin V/PI co-staining it is possible to discriminate among early-apoptotic (Annexin V+/PI–), late apoptotic/secondary necrotic (Annexin V+/PI+) or necrotic (PI+) cells (Fig. 2E) [31]. Semiquantitative analysis of the percentage of stained cells at different stages of death was performed (Fig. 2E). All cell types were analyzed before AA-treatment as controls. Virtually no PI and < 5% Annexin V stained cells were detected.
–
For GLU-WT cells, notwithstanding the high percentage of cells in the early-apoptotic stage at 90 min (80%), the percentage of necrotic and late-apoptotic cells was 75% and 15%, respectively, at 150 min, when cell viability is virtually lost (see Fig. 2A). In the case of RAF-WT cells, which evaded AA-PCD but did not grow, a percentage of early-apoptotic cells was still detectable at 90 min (65%), while the number of necrotic cells decreased to 15% and that of late-apoptotic cells were 70% at 150 min, when cell viability is 100%, in fairly good agreement with the number of cells with DNA fragmentation (see Fig. 2D). The percentage of Δrtg2 cells in raffinose at different stages of death was found to be similar to that of RAF-WT cells with almost no necrotic cells, whereas CAT8 or ADR1 deletion resulted in the virtual restoration of the necrotic phenotype of dead GLU-WT cells at 150 min, when all three knock-out cells showed 40-50% viability. Interestingly enough, RTG2 deletion in either Δcat8 or Δadr1 cells caused only apoptosis-like behaviour and no necrotic cell death at 150 min with about 70% cell viability. Then, since AA-PCD cells have been shown to accumulate ROS early en route to death, we also measured ROS after 30 min of acetic-acid treatment (Fig. 2F) [32]. GLU-WT cells showed the highest level (66%) of ROS, with 15% in RAF-WT cells and 55% in Δrtg2 cells in raffinose showing oxidative stress, in fairly good agreement with [16]. Δadr1 and Δcat8 cells, which lost viability similarly to Δrtg2 cells but with more necrotic cells, showed a lower level of ROS accumulation at 30 min (30% and 22%, respectively) (cf Fig. 2E and F). Δadr1Δrtg2 and Δcat8Δrtg2 cells, which behaved more like RAF-WT cells (with no necrotic cells) showed the lowest ROS level (18% and 9%, respectively).
–
Taken together, these data show that similar to RTG2, ADR1 and CAT8 also contribute to AA-PCD evasion under glucose de-repressing conditions, even though RTG2 deletion showed a different effect in WT or Δadr1/Δcat8 cells. Partial recovery of the ability of cells to evade AA-PCD in Δcat8Δrtg2 and Δadr1Δrtg2 double mutant cells showed unexpectedly that RTG2 is a suppressor of either CAT8 or ADR1 deletion. To get an insight into the mechanism of RTG2 suppression, we prepared Δadr1Δcat8Δrtg2, Δcat8Δmks1 and Δadr1Δmks1 cells and analyzed their viability after 200 min acetic acid treatment. Either the triple knock-out cells or the double knock-out cells, with constitutive activation of the RTG pathway, restored a WT phenotype (97%, 87% and 81% viability, respectively, at 200 min) like the double knock-out cells with the RTG pathway inactivated (Fig. 1S). Altogether these data suggest the Adr1 and Cat8 interact with Rtg2 and with each other in inducing cell resistance to AA-PCD in raffinose. In the absence of Adr1 and Cat8, AA-PCD evasion is acquired through activation of an alternative factor/pathway repressed by RTG2.
–
RTG pathway activation depends on ADR1 and CAT8 in raffinose-grown cells
Since it has previously been shown that RAF-WT evades AA-PCD by activating the RTG pathway, to gain insights into interaction between ADR1/CAT8 and the RTG pathway we analyzed mRNA expression of CIT2, whose up-regulation is a marker of RTG pathway activation [17], in Δrtg2, Δadr1, Δcat8, Δadr1Δrtg2, Δcat8Δrtg2 mutant strains along with WT controls. As a preliminary, CIT2 mRNA level was measured in mutant strains in exponential growth in raffinose as well as in GLU- and RAF-WT cells, as controls (Fig. 3A). The CIT2 mRNA level in GLU-WT cells was found to be ~13-fold lower than that in RAF-WT cells, which was taken as 100%, as previously found [18]. Both ADR1 and CAT8 deletion caused a ~4-fold reduction in the CIT2 mRNA level in raffinose, as compared with RAF-WT. A complete abolishment of CIT2 expression was found in Δrtg2, Δadr1Δrtg2 and Δcat8Δrtg2 mutant cells as expected. We then analyzed activation of RTG pathway over time in raffinose-grown WT and mutant cells with or without acetic acid treatment (Fig. 3B). GLU-WT cells en route to AA-PCD were also analyzed for comparison. A 3-4-fold CIT2 up-regulation was found in acetic acid-treated RAF-WT cells with respect to non-treated control samples at all time points analyzed, indicating RTG pathway activation in fairly good agreement with our published result [16]. In Δadr1 mutant cells, acetic acid treatment had little effect on CIT2 mRNA levels over the 200-min period (compare Δadr1 ctrl with Δadr1 AA in Fig. 3B). Importantly, CIT2 mRNA levels were lower in Δadr1 mutant cells than in acetic acid-treated, RAF-WT cells (compare Δadr1 AA with RAF-WT AA in Fig. 3B), suggesting that Δadr1 reduces cell ability to evade AA-PCD in raffinose medium possibly by limiting the activation of the RTG pathway. Similarly, no activation of the RTG pathway occurred in GLU-WT cells en route to AA-PCD, as previously reported [16].
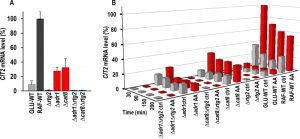 | FIGURE 3: CIT2 mRNA level in raffinose-grown wild-type and mutant strains in exponential phase growth and en route to acetic acid treatment. (A) CIT2 mRNA levels were measured by real-time PCR in RAF-WT cells and in deletion mutant cells grown in raffinose medium and collected in exponential phase (OD600 0.6-0.7). GLU-WT cells were also analyzed as control. Percentage of CIT2 mRNA levels, normalized to ACT1 mRNA, as compared with that in RAF-WT cells (100%), was reported. (B) CIT2 mRNA levels were measured by real-time PCR at the indicated time points in wild-type and mutant strains grown in raffinose medium with (AA, red bars) or without (ctrl, grey bars) the supplementation of 80 mM acetic acid. GLU-WT samples were included as controls. Percentage of CIT2 mRNA levels, normalized to ACT1 mRNA, as compared with that in AA-treated RAF-WT cells at 30 min, was reported. |
Interestingly, in Δcat8 cells that undergo AA-PCD to a similar extent as in Δadr1 cells, CIT2 mRNA levels were 2-3-fold higher in acetic acid-treated samples compared to untreated ones at all time points (compare Δcat8 AA with Δcat8 control in Fig. 3B), although the increase was at a lower degree than what was observed in RAF-WT cells. CIT2 expression was completely abolished in Δrtg2, Δadr1 Δrtg2 and Δcat8Δrtg2 cells either during AA treatment or in controls over the 200-min treatment period. The expression of DLD3, another RTG-target gene [33], was also measured. During exponential cell growth, differently from CIT2, DLD3 mRNA level in GLU-WT cells was 2-fold higher than in RAF-WT cells, but its expression, although strictly requiring RTG2, is neither regulated by ADR1 or CAT8 (Fig. 2SA) nor by acetic acid treatment in de-repression conditions (Fig. 2SB). Notwithstanding RTG pathway inactivation, AA-PCD evasion is observed in Δadr1Δrtg2 and Δcat8Δrtg2 double mutants differently from Δrtg2 or Δadr1 and Δcat8 single mutants, in which a low CIT2 expression was detected (Figure 3B). This suggests that activation of the RTG pathway is not the sole factor leading to AA-PCD resistance in RAF-WT cells. Rather, Δadr1Δrtg2 and Δcat8Δrtg2 double mutant strains acquired AA-PCD resistance through rewiring of other pathways.
DISCUSSION
Mitochondrial retrograde signaling has been shown to be dependent on the carbon source [34] and to cross-talk with other signaling pathways, including the CCR pathway, in response to mitochondrial dysfunction [22]. In this study we analyzed the relations between these two signaling pathways in the cell fate decision of yeast cells in response to acetic acid stress. We found that Adr1 and Cat8, two transcription factors dependent on Snf1, which regulates physiological reprogramming dependent on carbon-source availability, are strictly required for the activation of RTG2-dependent transcription in respiratory de-repression conditions, both during exponential growth, as judged by the strong CIT2 mRNA down-regulation found in both single mutants (Fig. 3A), and in AA-treated cells, in which deletion of either transcription factor causes increased cell death and down-regulation of CIT2 expression with respect to RAF-WT cells, which evade AA-PCD (cf Figs. 2C, D and 3B). Thus, in RAF-WT cells AA-PCD evasion is directly dependent on mitochondrial RTG pathway activation, as judged by the expression of CIT2, the prototypical RTG-target gene.
–
The observed difference between the expression of two RTG2-regulated genes, CIT2 and DLD3, shows how RTG-dependent mitochondrial retrograde signaling and the ensuing set of transcriptionally reprogrammed genes is strain-dependent. Indeed, DLD3 expression was not induced in ρ0 cells of the same yeast strain used in this study [33]. These data suggest that both ADR1 and CAT8 have a role in the determination of the up-regulated RTG-target genes and that Cat8/Adr1 and Rtg2 may regulate CIT2 expression independently. Here we found that in cells lacking either Adr1 or Cat8, both RTG2 and RTG3 deletion caused the same phenotype, i.e. restoration of AA-PCD evasion similar to RAF-WT cells (Figs. 2C and 1S). This, together with the observation that Δrtg2 and Δrtg3 cells undergo AA-PCD in a similar way, as shown here and in [16], shows that Cat8/Adr1 may regulate RTG-target gene expression directly or indirectly (Fig. 4).
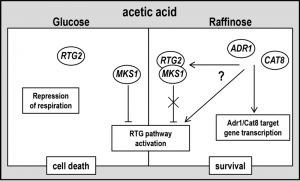 | FIGURE 4: A working hypothesis model for yeast acetic acid-stress response regulation by RTG and CCR pathways. Exponential glucose-grown cells undergo PCD in response to acetic acid with simultaneous repression of respiration and of RTG pathway. Raffinose-grown cells evade AA-PCD with de-repressed respiration [16] and RTG pathway activation. ADR1 and CAT8 are required for RTG pathway activation through an unknown mechanism (for details see text), showing a key role of SNF1-dependent CCR pathway in carbon source-regulation of mitochondrial RTG pathway. RTG2 may play a function in promoting cell death in repressing conditions when RTG pathway is inactive. |
In this respect, our results shed new light on the physiological role of RTG pathway activation in the evasion on AA-induced cell death. In fact, the RTG-pathway seems to have different functions in WT and in Δadr1 or Δcat8 cells in de-repression conditions. Although loss of viability in Δadr1 and Δcat8 cells was comparable to that observed with RTG2-lacking cells (Fig. 2A, B and [16]), the effects of RTG2 or ADR1/CAT8 deletion on the nature of the cell death process were different, as judged by the analysis of some morphological and biochemical features of AA-induced cell death. Acetic acid appeared to induce both an apoptosis-like PCD and a necrotic death process in GLU-WT cells. In raffinose de-repressing conditions, activation of the RTG pathway induced evasion of cell death in response to acetic acid, but with a strong decrease only in necrotic cell death. RTG2 deletion virtually abolished the necrotic phenotype while ADR1/CAT8 deletion decreased apoptosis-like PCD and increased necrosis. Moreover, in cells with functional ADR1 and CAT8 ROS level seemed to be highly dependent on RTG pathway activation, whereas in Δadr1 and Δcat8 cells, which seemingly have a lower mitochondrial activity, the ROS level was always significantly decreased and apparently independent on the RTG pathway (cf Figs. 2E, F and 3B). Altogether these data show that in respiratory-de-repressed raffinose-grown WT cells RTG pathway activation triggers an anti-oxidant, pro-survival cell response to acetic acid stress decreasing necrotic cell death. On the other hand, when the SNF1-axis of CCR pathway is inactivated by ADR1 or CAT8 deletion, RTG-dependent signaling causes a metabolic reprogramming leading to cell death. Thus genetic inactivation of the RTG pathway in Δadr1 or Δcat8 cells can restore AA-PCD resistance. Since it has been shown that Rtg2 controls RTG signaling by reversibly binding the negative regulator, Mks1 [35], it is tempting to speculate that Rtg2 may play a so far uncharacterized function in promoting (or repressing evasion from) necrotic cell death in glucose repressing condition, when RTG pathway is off, e.g. by dynamic interaction with an unknown factor/pathway.
–
Then, differently from in WT cells, AA-PCD evasion cannot be attributed to RTG pathway activation in the double mutants Δadr1Δrtg2 or Δcat8Δrtg2, since CIT2 expression is completely abolished in these cells, independent of acetic acid treatment (Figs. 2C, D and 3B). Thus the protective effect of raffinose on yeast AA-PCD likely requires also a SNF1-independent signaling pathway sensitive to carbon source or mitochondrial function. Since CAT8 and ADR1 co-regulate several glucose-sensitive genes and can act independently or synergistically [28][36][37], it could also be hypothesized that they can substitute for each other in the metabolic reprogramming of Δadr1Δrtg2 and Δcat8Δrtg2 cells in evading AA-PCD. This was excluded by our results since the triple knock-out cells Δadr1Δcat8Δrtg2 behave in the same way as the double knock-out cells, strongly suggesting the activation of alternative pathway/s inducing AA-PCD resistance.
–
We have previously shown that AA-PCD evasion occurs only in metabolic conditions characterized by concomitant activation of the RTG pathway and relief of CCR [16]. This study confirms that this is the case as demonstrated by the lack of effect on cell survival and DNA fragmentation upon genetic inactivation of CCR by deletion of HXK2 or MIG1 in glucose-grown cells (Fig. 2A, B), in which the RTG pathway is not activated by AA-treatment (Fig. 2E, F). This is in agreement with our previous observations that shift in glucose concentration from 2% to 0.5% can induce evasion of AA-PCD only in Δmks1 cells, in which the RTG pathway is constitutively activated [16]. Consistent with this, we found that Δmig1Δmks1 and Δhxk2Δmks1 did not acquire AA-PCD evasion in 2% glucose (Fig. 1S). This is because neither HXK2 nor MIG1 deletion phenocopies glucose de-repression, which occurs in RAF-WT or 0.5% glucose-grown cells. Moreover, the deletion of HAP4, whose transcription is under the negative control of the Mig1/Hxk2 complex, did not abolish AA-PCD evasion in raffinose-grown cells, suggesting that the Mig1/Hxk2 axis of CCR does not significantly contribute to glucose-dependent control of yeast acetic acid-stress sensitivity. It is of note that enhancement of AA-PCD by deletion of HXK2 has been found in a different yeast strain [38]. This could be due in part to the different degree of activation of CCR and the cAMP/PKA signaling pathway in different genetic background and growth conditions [39].
–
Thus, metabolic reprogramming caused by concomitant activation of the RTG pathway and SNF1-dependent relief of CCR appears to be required for AA-PCD evasion. Genome-wide transcriptome analyses have shown both that RTG2-dependent genes are involved in peroxisomal biogenesis and anaplerotic reactions of tricarboxylic acid cycle intermediates, directing carbon metabolism to α-ketoglutarate biosynthesis [40], and that ADR1-dependent genes channel metabolites into acetyl-CoA production [28]. Yeast cells grown on glucose cannot metabolize acetic acid due to activation of CCR ([41] and refs therein) explaining in part why yeast is sensitive to acetic acid stress in the presence of glucose. Yet, our data show that relief of CCR, without up-regulation of RTG-target genes, is not sufficient to induce AA-PCD evasion. Intracellular acetate is transformed into acetyl-CoA through ACS1 and ACS2, which can be used as a metabolite for metabolic pathways or a substrate for lysine acetyltransferases in epigenetic regulation of gene expression through histone acetylation [42]. In view of the role of RTG2, ADR1 and CAT8 in chromatin remodeling [36][43] it is tempting to speculate that simultaneous activation of the RTG pathway and SNF1/AMPK-target gene expression can signal metabolic control of AA-PCD through modulation of acetyl-CoA levels, as already shown for respiratory-deficient ageing yeast cells [44]. Our data confirm the use of yeast as a model for a better understanding of cell adaptations to the extracellular environment and revealing the molecular basis of PCD evasion in eukaryotes.
MATERIALS AND METHODS
Yeast strains, growth conditions and acetic acid treatment
The S. cerevisiae strains used in this study are listed in Table 1. Cells were grown at 30°C in YPD or YPR (1% yeast extract, 2% bactopeptone, and 2% glucose or raffinose, respectively). Acetic acid treatment was carried out as described [45]. Briefly, cells were grown at 26°C up to exponential phase (OD600 = 0.6 – 0.8) in YPD or YPR, resuspended (107 cells/ml) in the same medium adjusted to pH 3.00 with HCl, with or without the supplementation of 80 mM acetic acid and incubated for different times at 26°C. Cell viability was determined by measuring colony forming units (cfu) after 2 days of growth on YPD plates at 30°C.
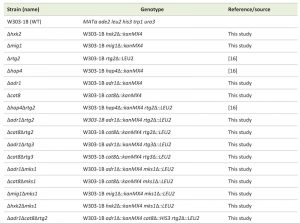 | TABLE 1. Strains of Saccharomyces cerevisiae used in this study. [16] |
Mutant strain construction
To delete ADR1, CAT8, HXK2, and MIG1 genes in W303-1B, disruption cassettes with the kanMX4 selection marker were amplified by PCR using genomic DNA from respective deletion mutant strains in the BY4741 strain background (Yeast genome deletion project) as template. PCR products of the disruption cassettes were transformed into W303-1B strain using the high efficiency yeast transformation method [46]. Gene disruption was confirmed by PCR genotyping. To generate Δadr1Δrtg2 and Δcat8Δrtg2 double mutant strains, a plasmid carrying an rtg2::LEU2 disruption cassette (pUCrtg2::LEU2, [47]) was digested with PstI and transformed into Δadr1 or Δcat8 single knock-out strains. To generate Δadr1Δrtg3 and Δcat8 Δrtg3 strains, a Δrtg3::LEU2 cassette was amplified from Δrtg3 cells [16] using the primer pair (F) 5’- CGAAAGTGAGGCTGAGAACC-3’ and (R) 5’-GACTCTCCATAGTG-CCAGCA-3’ and transformed into Δadr1 or Δcat8 single knock-out strains. Δrtg2 and Δrtg3 mutations were confirmed by the glutamate auxotrophy phenotype and PCR genotyping. For generating the Δcat8Δadr1Δrtg2 strain, we first cloned pBS-Δcat8::kanmX4 and then replaced the kanMX4 cassette with a HIS3 cassette. pBS-cat8::HIS3 was used to transform Δadr1Δrtg2 knock-out cells. To generate Δadr1Δmks1, Δcat8Δmks1, Δmig1Δmks1 and Δhxk2Δmks1 double mutants, a Δmks1::LEU2 disruption cassette was introduced into respective single mutant strains and transformants were selected on minimal medium lacking leucine. Δmks1::LEU2 mutations were confirmed by PCR-genotyping.
–
TUNEL assay, Annexin V/PI staining and ROS detection
DNA fragmentation was detected by TUNEL assay as reported in [16]. Briefly, acetic acid-treated and untreated control cells (2 x 107) were harvested at the 150-min time point, fixed in 3.7% formaldehyde solution in PBS, digested with 750 μg/ml zymolyase 20T and incubated in permeabilization solution (0.1% Triton-X100, 0.1% sodium citrate) for 2 min on ice, and then 30 μl TUNEL reaction mixture was added (In Situ Cell Death Detection kit, Fluorescein, Roche) for 1 hour at 37°C. After incubation cells were washed, resuspended in PBS and observed using a Leica TCS SP5 confocal microscope. Plasma membrane integrity was measured by propidium iodide (PI) staining essentially as described in [48]. Briefly, 1 x 107 cells were centrifuged at 10,000 g for 3 min and resuspended in 10 μl medium, treated with 0.5 μl PI (500 μg/ml), incubated for 15 min at room temperature in the dark and observed with a confocal microscope (see below). PS exposure on the cell surface and membrane integrity were detected by a fluorescein isothiocyanate (FITC)-coupled Annexin V reaction, using the Annexin-V-FLUOS Staining Kit (Roche). AA-treated and control yeast cells (2 x 107) were sedimented at 10,000 g for 3 min at different times and digested with 750 μg/ml zymolyase 20T in sorbitol buffer (1.2 M sorbitol, 0.5 mM MgCl2, 35 mM potassium phosphate pH 6.8) at 30°C for 1 h. Cells were washed twice with binding buffer (10 mM HEPES/NaOH pH 7.4, 140 mM NaCl, 2.5 mM CaCl2, 1.2 M sorbitol) and 30 μl label solution (2 μl Annexin V, 2 μl PI, 98 μl binding buffer) were added to 50 μl cell suspension (1 x 107) for 20 min incubation in the dark at room temperature. After washing, the cells were applied to microscopic slides and observed using LEICA TCS SP5 confocal microscopy (HCX PL APO lambda blue 63 x 1.40 objective) exciting the sample with a Argon Laser at 488 nm and emission at 494 – 537 nm, for FITC-Annexin V, and at 629 – 776 nm, for PI. Digital images were analyzed using LAS X software. To detect intracellular ROS levels, 10 μg/ml 2’,7-dichlorofluorescein diacetate (H2DCF-DA; Molecular Probes) dissolved in ethanol was added to cells both 30 min before and during cell treatment with or without AA. AA-treated or control cells were harvested at different times and oxidation to the fluorophore dichlorofluorescein (DCF) was detected by confocal fluorescence microscopy analysis as above, with excitation at 488 nm and emission at 500-600 nm.
–
Real-time polymerase chain reaction (PCR)
CIT2 and DLD3 mRNA levels were determined in acetic acid-treated or control cells collected at the exponential phase (OD600 = 0.7). 20 ml of cell cultures were withdrawn at different times and centrifuged at 3000 g. Cells were either stored at −80°C or immediately lysed with by zymolyase 20T to extract total RNA using Presto™ Mini RNA Yeast Kit (Geneaid Biotech Ltd). 1 μg RNA (OD260/OD280 ≥ 1.9) was immediately reverse-transcribed using QuantiTect® Reverse Transcription Kit (Qiagen) and cDNA used for real-time PCR analysis or stored at −20°C. Real-time PCR was carried out using a QuantiTect® SYBR Green PCR Kit (Qiagen) on an Applied Biosystems QuantStudio™ 6 Flex machine using the following primer pairs: for CIT2: (F) 5′-CGGTTATGGTCATGCTGTGCT-3′ and (R) 5′- GGTCCATGGCAAACTTACGCT-3′; for ACT1: (F) 5′-CTTTGGCT-CCATCTTCCATG-3′ and (R) 5′-CACCAATCCAGACGGAGTACTT-3′; for DLD3 see [49]. The fold-increase (2-ΔΔCt) of CIT2 and DLD3 mRNA levels, normalized to ACT1 mRNA, as compared with that in RAF-WT cells was calculated and reported as the percentage of RAF-WT CIT2 mRNA level taken as 100%.
References
- H.G. Crabtree, "Observations on the carbohydrate metabolism of tumours", Biochemical Journal, vol. 23, pp. 536-545, 1929. http://dx.doi.org/10.1042/bj0230536
- R. Lagunas, "Energetic irrelevance of aerobiosis for S. cerevisiae growing on sugars", Molecular and Cellular Biochemistry, vol. 27, pp. 139-146, 1979. http://dx.doi.org/10.1007/bf00215362
- J.M. Gancedo, "Yeast carbon catabolite repression.", Microbiology and molecular biology reviews : MMBR, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9618445
- F. ROLLAND, J. WINDERICKX, and J. THEVELEIN, "Glucose-sensing and -signalling mechanisms in yeast", FEMS Yeast Research, vol. 2, pp. 183-201, 2002. http://dx.doi.org/10.1016/s1567-1356(02)00046-6
- H. Schüller, "Transcriptional control of nonfermentative metabolism in the yeast Saccharomyces cerevisiae.", Current genetics, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12715202
- �. Kayikci, and J. Nielsen, "Glucose repression inSaccharomyces cerevisiae", FEMS Yeast Research, vol. 15, pp. fov068, 2015. http://dx.doi.org/10.1093/femsyr/fov068
- M. Johnston, and J. Kim, "Glucose as a hormone: receptor-mediated glucose sensing in the yeast Saccharomyces cerevisiae", Biochemical Society Transactions, vol. 33, pp. 247-252, 2005. http://dx.doi.org/10.1042/bst0330247
- A. Kaniak, Z. Xue, D. Macool, J. Kim, and M. Johnston, "Regulatory Network Connecting Two Glucose Signal Transduction Pathways in Saccharomyces cerevisiae", Eukaryotic Cell, vol. 3, pp. 221-231, 2004. http://dx.doi.org/10.1128/ec.3.1.221-231.2004
- S. Ozcan, and M. Johnston, "Function and regulation of yeast hexose transporters.", Microbiology and molecular biology reviews : MMBR, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10477308
- M. Carlson, "Glucose repression in yeast", Current Opinion in Microbiology, vol. 2, pp. 202-207, 1999. http://dx.doi.org/10.1016/s1369-5274(99)80035-6
- K. Hedbacker, "SNF1/AMPK pathways in yeast", Frontiers in Bioscience, vol. 13, pp. 2408, 2008. http://dx.doi.org/10.2741/2854
- E. Boles, and C.P. Hollenberg, "The molecular genetics of hexose transport in yeasts", FEMS Microbiology Reviews, vol. 21, pp. 85-111, 1997. http://dx.doi.org/10.1111/j.1574-6976.1997.tb00346.x
- B. Smets, R. Ghillebert, P. De Snijder, M. Binda, E. Swinnen, C. De Virgilio, and J. Winderickx, "Life in the midst of scarcity: adaptations to nutrient availability in Saccharomyces cerevisiae", Current Genetics, vol. 56, pp. 1-32, 2010. http://dx.doi.org/10.1007/s00294-009-0287-1
- N. Guaragnella, L. Antonacci, S. Passarella, E. Marra, and S. Giannattasio, "Achievements and perspectives in yeast acetic acid-induced programmed cell death pathways", Biochemical Society Transactions, vol. 39, pp. 1538-1543, 2011. http://dx.doi.org/10.1042/bst0391538
- N. Guaragnella, M. Ždralević, L. Antonacci, S. Passarella, E. Marra, and S. Giannattasio, "The role of mitochondria in yeast programmed cell death", Frontiers in Oncology, vol. 2, 2012. http://dx.doi.org/10.3389/fonc.2012.00070
- N. Guaragnella, M. Ždralević, P. Lattanzio, D. Marzulli, T. Pracheil, Z. Liu, S. Passarella, E. Marra, and S. Giannattasio, "Yeast growth in raffinose results in resistance to acetic-acid induced programmed cell death mostly due to the activation of the mitochondrial retrograde pathway", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1833, pp. 2765-2774, 2013. http://dx.doi.org/10.1016/j.bbamcr.2013.07.017
- Z. Liu, and R.A. Butow, "Mitochondrial Retrograde Signaling", Annual Review of Genetics, vol. 40, pp. 159-185, 2006. http://dx.doi.org/10.1146/annurev.genet.40.110405.090613
- P.A. Kirchman, S. Kim, C.Y. Lai, and S.M. Jazwinski, "Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae.", Genetics, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10224252
- D. Journo, A. Mor, and H. Abeliovich, "Aup1-mediated Regulation of Rtg3 during Mitophagy", Journal of Biological Chemistry, vol. 284, pp. 35885-35895, 2009. http://dx.doi.org/10.1074/jbc.m109.048140
- P. Spincemaille, N. Matmati, Y.A. Hannun, B.P. Cammue, and K. Thevissen, "Sphingolipids and mitochondrial function in budding yeast", Biochimica et Biophysica Acta (BBA) - General Subjects, vol. 1840, pp. 3131-3137, 2014. http://dx.doi.org/10.1016/j.bbagen.2014.06.015
- Y. Jia, B. Rothermel, J. Thornton, and R.A. Butow, "A Basic Helix-Loop-Helix–Leucine Zipper Transcription Complex in Yeast Functions in a Signaling Pathway from Mitochondria to the Nucleus", Molecular and Cellular Biology, vol. 17, pp. 1110-1117, 1997. http://dx.doi.org/10.1128/mcb.17.3.1110
- Z. Liu, and R.A. Butow, "A Transcriptional Switch in the Expression of Yeast Tricarboxylic Acid Cycle Genes in Response to a Reduction or Loss of Respiratory Function", Molecular and Cellular Biology, vol. 19, pp. 6720-6728, 1999. http://dx.doi.org/10.1128/mcb.19.10.6720
- J. Blom, M.J.T. De Mattos, and L.A. Grivell, "Redirection of the Respiro-Fermentative Flux Distribution in Saccharomyces cerevisiae by Overexpression of the Transcription Factor Hap4p", Applied and Environmental Microbiology, vol. 66, pp. 1970-1973, 2000. http://dx.doi.org/10.1128/aem.66.5.1970-1973.2000
- S.L. Forsburg, and L. Guarente, "Identification and characterization of HAP4: a third component of the CCAAT-bound HAP2/HAP3 heteromer.", Genes & Development, vol. 3, pp. 1166-1178, 1989. http://dx.doi.org/10.1101/gad.3.8.1166
- I. Bourges, M. Mucchielli, C.J. Herbert, B. Guiard, G. Dujardin, and B. Meunier, "Multiple Defects in the Respiratory Chain Lead to the Repression of Genes Encoding Components of the Respiratory Chain and TCA Cycle Enzymes", Journal of Molecular Biology, vol. 387, pp. 1081-1091, 2009. http://dx.doi.org/10.1016/j.jmb.2009.02.039
- S. Buschlen, J. Amillet, B. Guiard, A. Fournier, C. Marcireau, and M. Bolotin-Fukuhara, "TheS. cerevisiaeHAP Complex, a Key Regulator of Mitochondrial Function, Coordinates Nuclear and Mitochondrial Gene Expression", Comparative and Functional Genomics, vol. 4, pp. 37-46, 2003. http://dx.doi.org/10.1002/cfg.254
- R. Lascaris, H.J. Bussemaker, A. Boorsma, M. Piper, H. van der Spek, L. Grivell, and J. Blom, "Hap4p overexpression in glucose-grown Saccharomyces cerevisiae induces cells to enter a novel metabolic state", Genome Biology, vol. 4, 2002. http://dx.doi.org/10.1186/gb-2002-4-1-r3
- E.T. Young, K.M. Dombek, C. Tachibana, and T. Ideker, "Multiple Pathways Are Co-regulated by the Protein Kinase Snf1 and the Transcription Factors Adr1 and Cat8", Journal of Biological Chemistry, vol. 278, pp. 26146-26158, 2003. http://dx.doi.org/10.1074/jbc.m301981200
- V. Haurie, M. Perrot, T. Mini, P. Jenö, F. Sagliocco, and H. Boucherie, "The Transcriptional Activator Cat8p Provides a Major Contribution to the Reprogramming of Carbon Metabolism during the Diauxic Shift in Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 276, pp. 76-85, 2001. http://dx.doi.org/10.1074/jbc.m008752200
- B. Turcotte, X.B. Liang, F. Robert, and N. Soontorngun, "Transcriptional regulation of nonfermentable carbon utilization in budding yeast", FEMS Yeast Research, vol. 10, pp. 2-13, 2010. http://dx.doi.org/10.1111/j.1567-1364.2009.00555.x
- D. Carmona-Gutierrez, T. Eisenberg, S. Büttner, C. Meisinger, G. Kroemer, and F. Madeo, "Apoptosis in yeast: triggers, pathways, subroutines", Cell Death & Differentiation, vol. 17, pp. 763-773, 2010. http://dx.doi.org/10.1038/cdd.2009.219
- N. Guaragnella, L. Antonacci, S. Passarella, E. Marra, and S. Giannattasio, "Hydrogen peroxide and superoxide anion production during acetic acid-induced yeast programmed cell death", Folia Microbiologica, vol. 52, 2007. http://dx.doi.org/10.1007/bf02931304
- A. Chelstowska, Z. Liu, Y. Jia, D. Amberg, and R.A. Butow, "Signalling between mitochondria and the nucleus regulates the expression of a new D-lactate dehydrogenase activity in yeast.", Yeast (Chichester, England), 1999. http://www.ncbi.nlm.nih.gov/pubmed/10509019
- R.A. Butow, and N.G. Avadhani, "Mitochondrial Signaling", Molecular Cell, vol. 14, pp. 1-15, 2004. http://dx.doi.org/10.1016/s1097-2765(04)00179-0
- Z. Liu, T. Sekito, M. Špı́rek, J. Thornton, and R.A. Butow, "Retrograde Signaling Is Regulated by the Dynamic Interaction between Rtg2p and Mks1p", Molecular Cell, vol. 12, pp. 401-411, 2003. http://dx.doi.org/10.1016/s1097-2765(03)00285-5
- R.K. Biddick, G.L. Law, and E.T. Young, "Adr1 and Cat8 Mediate Coactivator Recruitment and Chromatin Remodeling at Glucose-Regulated Genes", PLoS ONE, vol. 3, pp. e1436, 2008. http://dx.doi.org/10.1371/journal.pone.0001436
- C. Tachibana, J.Y. Yoo, J. Tagne, N. Kacherovsky, T.I. Lee, and E.T. Young, "Combined Global Localization Analysis and Transcriptome Data Identify Genes That Are Directly Coregulated by Adr1 and Cat8", Molecular and Cellular Biology, vol. 25, pp. 2138-2146, 2005. http://dx.doi.org/10.1128/mcb.25.6.2138-2146.2005
- L. Amigoni, E. Martegani, and S. Colombo, "Lack of HXK2 induces localization of active Ras in mitochondria and triggers apoptosis in the yeast Saccharomyces cerevisiae.", Oxidative medicine and cellular longevity, 2013. http://www.ncbi.nlm.nih.gov/pubmed/24089630
- A. Kümmel, J.C. Ewald, S. Fendt, S.J. Jol, P. Picotti, R. Aebersold, U. Sauer, N. Zamboni, and M. Heinemann, "Differential glucose repression in common yeast strains in response to HXK2 deletion", FEMS Yeast Research, vol. 10, pp. 322-332, 2010. http://dx.doi.org/10.1111/j.1567-1364.2010.00609.x
- C.B. Epstein, J.A. Waddle, W. Hale, V. Davé, J. Thornton, T.L. Macatee, H.R. Garner, and R.A. Butow, "Genome-wide Responses to Mitochondrial Dysfunction", Molecular Biology of the Cell, vol. 12, pp. 297-308, 2001. http://dx.doi.org/10.1091/mbc.12.2.297
- S. Giannattasio, N. Guaragnella, M. Ždralević, and E. Marra, "Molecular mechanisms of Saccharomyces cerevisiae stress adaptation and programmed cell death in response to acetic acid", Frontiers in Microbiology, vol. 4, 2013. http://dx.doi.org/10.3389/fmicb.2013.00033
- L. Galdieri, T. Zhang, D. Rogerson, R. Lleshi, and A. Vancura, "Protein Acetylation and Acetyl Coenzyme A Metabolism in Budding Yeast", Eukaryotic Cell, vol. 13, pp. 1472-1483, 2014. http://dx.doi.org/10.1128/ec.00189-14
- M.G. Pray-Grant, D. Schieltz, S.J. McMahon, J.M. Wood, E.L. Kennedy, R.G. Cook, J.L. Workman, J.R. Yates, and P.A. Grant, "The Novel SLIK Histone Acetyltransferase Complex Functions in the Yeast Retrograde Response Pathway", Molecular and Cellular Biology, vol. 22, pp. 8774-8786, 2002. http://dx.doi.org/10.1128/mcb.22.24.8774-8786.2002
- R. Friis, J. Glaves, T. Huan, L. Li, B. Sykes, and M. Schultz, "Rewiring AMPK and Mitochondrial Retrograde Signaling for Metabolic Control of Aging and Histone Acetylation in Respiratory-Defective Cells", Cell Reports, vol. 7, pp. 565-574, 2014. http://dx.doi.org/10.1016/j.celrep.2014.03.029
- S. Giannattasio, N. Guaragnella, M. Corte-Real, S. Passarella, and E. Marra, "Acid stress adaptation protects Saccharomyces cerevisiae from acetic acid-induced programmed cell death", Gene, vol. 354, pp. 93-98, 2005. http://dx.doi.org/10.1016/j.gene.2005.03.030
- D.C. Amberg, D.J. Burke, and J.N. Strathern, "High-efficiency transformation of yeast.", CSH protocols, 2006. http://www.ncbi.nlm.nih.gov/pubmed/22485546
- B.A. Rothermel, A.W. Shyjan, J.L. Etheredge, and R.A. Butow, "Transactivation by Rtg1p, a Basic Helix-Loop-Helix Protein That Functions in Communication between Mitochondria and the Nucleus in Yeast", Journal of Biological Chemistry, vol. 270, pp. 29476-29482, 1995. http://dx.doi.org/10.1074/jbc.270.49.29476
- F. Madeo, E. Fröhlich, and K. Fröhlich, "A Yeast Mutant Showing Diagnostic Markers of Early and Late Apoptosis", The Journal of Cell Biology, vol. 139, pp. 729-734, 1997. http://dx.doi.org/10.1083/jcb.139.3.729
- A.N. Starovoytova, M.I. Sorokin, S.S. Sokolov, F.F. Severin, and D.A. Knorre, "Mitochondrial signaling inSaccharomyces cerevisiaepseudohyphae formation induced by butanol", FEMS Yeast Research, vol. 13, pp. 367-374, 2013. http://dx.doi.org/10.1111/1567-1364.12039
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We thank Prof. Shawn Doonan for critical reading of the manuscript. This work has been funded by grants from the Italian Ministry of Economy and Finance to the CNR for the Project “FaReBio di Qualità”, project BioNet-PTP - PO Regione Puglia FESR 2000-2006 and FCRP project “Identificazione di molecole attive per lo sviluppo di nuovi farmaci antitumorali contro il carcinoma di prostata” to S.G. L.L. is recipient of a CNR research contract funded by the FCRP project.
COPYRIGHT
© 2016

The transcription factors ADR1 or CAT8 are required for RTG pathway activation and evasion from yeast acetic acid-induced programmed cell death in raffinose by Luna Laera et al. is licensed under a Creative Commons Attribution 4.0 International License.








