
Research Articles:
Microbial Cell, Vol. 10, No. 7, pp. 145 - 156; doi: 10.15698/mic2023.07.800
GFP fusions of Sec-routed extracellular proteins in Staphylococcus aureus reveal surface-associated coagulase in biofilms
1 Interdisciplinary Nanoscience Center, Aarhus University, Aarhus, Denmark.
2 Department of Physics, University of York, York, UK.
3 Department of Biochemistry and Molecular Biology, University of Southern Denmark, Odense, Denmark.
4 Department of Biology, University of York, York, UK.
5 Department of Biology, Aarhus University, Aarhus, Denmark.
# Joint first authors.
Keywords: fusion protein, Gram positive bacteria, monomeric superfolder GFP, coagulase, biofilms.
Abbreviations:
Coa – coagulase,
GFP – green fluorescent protein,
msfGFP – monomeric superfolder GFP,
Sec – Secretory pathway,
Tat – Twin Arginine Translocation pathway,
vWbp – von Willebrand factor binding protein.
Received originally: 22/12/2022 Received in revised form: 14/06/2023
Accepted: 19/06/2023
Published: 28/06/2023
Correspondence:
Rikke L. Meyer, Interdisciplinary Nanoscience Center, Aarhus University, Gustav Wieds Vej 14, 8000 Aarhus C, Denmark; rikke.meyer@inano.au.dk
Mark C. Leake, Departments of Physics and Biology, University of York, York, YO10 5DD, UK; mark.leake@york.ac.uk
Conflict of interest statement: The authors declare no conflicts of interest.
Please cite this article as: Dominique C. S. Evans, Amanda B. Khamas, Lisbeth Marcussen, Kristian S. Rasmussen, Janne K. Klitgaard, Birgitte H. Kallipolitis, Janni Nielsen, Daniel E. Otzen, Mark C. Leake and Rikke L. Meyer (2023). GFP fusions of Sec-routed extracellular proteins in Staphylococcus aureus reveal surface-associated coagulase in biofilms. Microbial Cell 10(7): 145-156. doi: 10.15698/mic2023.07.800
Abstract
Staphylococcus aureus is a major human pathogen that utilises many surface-associated and secreted proteins to form biofilms and cause disease. However, our understanding of these processes is limited by challenges of using fluorescent protein reporters in their native environment, because they must be exported and fold correctly to become fluorescent. Here, we demonstrate the feasibility of using the monomeric superfolder GFP (msfGFP) exported from S. aureus. By fusing msfGFP to signal peptides for the Secretory (Sec) and Twin Arginine Translocation (Tat) pathways, the two major secretion pathways in S. aureus, we quantified msfGFP fluorescence in bacterial cultures and cell-free supernatant from the cultures. When fused to a Tat signal peptide, we detected msfGFP fluorescence inside but not outside bacterial cells, indicating a failure to export msfGFP. However, when fused to a Sec signal peptide, msfGFP fluorescence was present outside cells, indicating successful export of the msfGFP in the unfolded state, followed by extracellular folding and maturation to the photoactive state. We applied this strategy to study coagulase (Coa), a secreted protein and a major contributor to the formation of a fibrin network in S. aureus biofilms that protects bacteria from the host immune system and increases attachment to host surfaces. We confirmed that a genomically integrated C-terminal fusion of Coa to msfGFP does not impair the activity of Coa or its localisation within the biofilm matrix. Our findings demonstrate that msfGFP is a good candidate fluorescent reporter to consider when studying proteins secreted by the Sec pathway in S. aureus.
INTRODUCTION
Green fluorescent protein (GFP) has been used for decades as an intracellular reporter for gene expression and as a fluorescent tag to visualise single proteins in the cytoplasm of bacteria [1]. An advantage of fluorescent proteins is that samples do not need to be stained and incubated to visualise the protein. GFP and other fluorescent proteins have therefore been instrumental for studies into protein localisation, visualising subcellular compartments, monitoring gene expression, tissue labelling, as well as DNA and RNA labelling [2].
–
While GFP fusion proteins have taught us much about intracellular proteins, little research has been done on extracellular proteins, such as surface-bound proteins or other secreted proteins. Some GFP variants have been successfully secreted to the periplasm and outer membrane of Gram-negative bacteria [3][4][5], however, there are only few examples of this for Gram-positive bacteria. To our knowledge, GFP secretion in Gram-positive bacteria has only been achieved in a small number of organisms, including Corynebacterium glutamicum [6], Bacillus subtilis [7][8], Streptococcus mutans [9], Mycobacterium smegmatis [10], and Staphylococcus epidermidis [11]. Split GFP has additionally been successfully secreted by B. subtilis [12]. There is a multitude of reasons why generation of GFP-fusion proteins may fail. In particular, the fusion protein may not be successfully secreted, the GFP may misfold and fail to become fluorescent in the extracellular environment, or the chromophore may not mature properly [6][13]. Additionally, the level of transcription and translation, protein turnover rate, and photobleaching further complicate imaging of GFP fusions [13].
–
Most extracellular proteins are secreted in an unfolded state via the Secretory (Sec) pathway, where they are exported across the cytosolic membrane into the periplasm in Gram-negative bacteria or outside the cell in Gram-positive bacteria [14]. It is a highly conserved pathway present in all classes of bacteria [15]. Sec-routed proteins have a signal peptide at their N-terminus that directs them towards the SecYEG membrane protein channel, after which they are driven stepwise across the membrane by the ATPase molecular motor SecA [16]. The transported protein then folds on the trans side of the membrane. In many Gram-negative bacteria, SecB stabilises and targets the unfolded protein to SecA, while in Gram-positive and other Gram-negative bacteria, general chaperones maintain the protein in an unfolded state [16]. Another common secretion pathway is the Twin Arginine Translocation (Tat) pathway, in which proteins are exported in a folded state [17], however, not all bacterial species have a Tat pathway [18]. Tat-routed proteins have an N-terminal signal sequence containing a twin-arginine motif that gives the pathway its name [19]. Some proteins that are secreted through the Tat-pathway, such as proteins with co-factors that bind to cytoplasmic proteins, usually need to fold in the cytoplasm to function correctly [20]. The Tat pathway contains three subunits TatA, TatB, and TatC in Gram-negative bacteria and two subunits TatA and TatC in Gram-positive bacteria, which bind the signal peptide and form a membrane spanning channel [15]. These subunits have been studied previously using fluorescent protein reporters in live Escherichia coli cells [21]. Folded proteins are exported outside of the cell in Gram-positive bacteria, and to the periplasm in Gram-negative bacteria, where they may be exported across the outer membrane via other mechanisms [15].
–
The aims of our present study were to investigate whether monomeric superfolder GFP (msfGFP) is a good candidate for extracellular fusion proteins in Staphylococcus aureus, and to determine if msfGFP can be secreted by either of the two secretion pathways Sec and Tat. S. aureus is a Gram-positive coccus which has both the Sec and Tat secretion pathways [15][18]. It is a major biofilm-forming human pathogen that can cause skin and soft tissue infections, endocarditis, osteomyelitis, and toxic shock syndrome [22]. S. aureus utilises many surface-associated and secreted proteins to interact with host tissue, to establish infections, and evade the immune system [23]. These proteins include a family known as microbial surface components recognising adhesive matrix molecules (MSCRAMMs), all of which contain a Sec signal peptide [23]. Examples include clumping factors A and B (ClfA and ClfB) that clump bacteria by binding host fibrinogen and aid tissue colonisation [24], fibronectin binding proteins A and B (FnBPA and FnBPB) that bind host fibronectin, fibrinogen, and elastin, and therefore facilitate attachment to host tissues via host proteins [24], and collagen adhesin (Cna) that facilitates attachment via collagen and helps S. aureus escape immune cells [23]. S. aureus also secretes a family of proteins called secretable expanded repertoire adhesive molecules (SERAMs). These include extracellular adherence protein (Eap), extracellular matrix protein-binding protein (Emp), extracellular fibrinogen binding protein (Efp), coagulase (Coa), and von Willebrand factor binding protein (vWbp). Eap inhibits neutrophils and therefore inhibits the immune response [25], Emp binds host fibronectin, fibrinogen, and vitronectin [26], which appears to be important for virulence [26], and Efp inhibits phagocytosis [27] and decreases wound healing [28]. Coa and vWbp bind to and activate host prothrombin to hijack the host coagulation cascade and thereby trigger the formation of fibrin fibers, a major component of the biofilm extracellular matrix [29], in two concentric structures: a cell surface-associated pseudocapsule and an extended outer network, which together act as mechanical barriers against immune attack [30], enhance virulence [31], and increase adhesion to surfaces [32]. S. aureus would benefit from a reliable system with which to label and visualise proteins such as these that are important to its virulence and pathogenicity, especially in complex environments such as biofilms where traditional antibody labelling methods may fail. Antibodies are approximately 10 nm in size [33], which is relatively large compared to many matrix components, such as DNA which has a width of approximately 2.5 nm and many proteins which are less than 10 nm in size. Therefore, antibodies may fail to penetrate some biofilm matrices and fail to label them correctly.
–
We chose msfGFP as our model fluorescent protein due to its brightness and enhanced folding properties [34], and it has been previously shown to fold in traditionally challenging environments such as the periplasm of Gram-negative bacteria [34]. We investigated Sec- and Tat-secreted msfGFP by fusing msfGFP to Sec and Tat signal peptides in overexpression plasmids and subsequently measuring the increase in fluorescence from bacterial cultures and cell-free culture supernatants. After confirming that msfGFP is suitable to visualise secreted proteins, we developed a C-terminal chromosome-integrated fusion between msfGFP and Coa in S. aureus, which is predicted to have a Sec-type signal peptide [35]. We demonstrated that fusion to msfGFP did not impair the biological function of Coa, and that Coa:msfGFP fusion proteins revealed the location of Coa in S. aureus biofilms. Coa is responsible for producing a fibrin pseudocapsule and has previously been located within the pseudocapsule [30][31]. We demonstrate that Coa localises to cell surfaces, where we hypothesise that it associates with the cell to facilitate fibrin production near the surface of bacteria.
RESULTS
msfGFP is secreted via Sec and becomes fluorescent in the extracellular environment
The fluorescent protein msfGFP is a good candidate for tagging extracellular proteins in Gram-positive bacteria, but its implementation depends on whether it can be secreted and fold properly in the extracellular space. We therefore tested the ability of msfGFP to become fluorescent after secretion via the Tat and Sec pathways in S. aureus. We generated four strains of S. aureus that carried different variants of the overexpression pRMC2 plasmid (Figure 1). Strain 1 contained an empty pRMC2 vector, which served as negative control, strain 2 contained pRMC2 encoding msfGFP without a signal peptide and was used as a positive control to verify msfGFP expression, strain 3 contained pRMC2 encoding Tat:msfGFP for secretion of GFP through the Tat pathway, and strain 4 contained pRMC2 encoding Sec:msfGFP for secretion through the Sec pathway. The presence of functional msfGFP was then measured as the appearance of green fluorescence of cultures and cell-free supernatants using a fluorescence plate reader after inducing expression of msfGFP from the plasmid.
–
 | FIGURE 1: Visual schematics of constructs expressing fusion proteins under control of the inducible Pxyl/tetO promoter in pRMC2. (A) Tat:msfGFP, (B) Sec:msfGFP, and (C) msfGFP control. SD = Shine-Dalgarno sequence, SP = signal peptide, and L = linker. Amino acid sequences for the Tat signal peptide [18], Sec signal peptide [45] and linker are given in the figure. DNA sequences are provided in Supplementary S1. |
–
Only the culture expressing Sec:msfGFP produced fluorescence in the cell-free supernatant, which indicated that msfGFP can secrete and fold correctly when exported by the Sec-pathway (Figure 2A). The fluorescence intensity from the culture (bacteria and supernatant) was at a similar level to the supernatant alone, indicating that msfGFP was primarily present in the supernatant. In cultures expressing Tat:msfGFP or msfGFP without a signal peptide, fluorescence was detected in bacterial cultures but not the supernatants (Figure 2A), indicating that msfGFP could fold correctly within cells, but was not secreted via the Tat pathway. Although Tat:msfGFP was not successfully secreted, the fluorescence intensity from Tat:msfGFP cell cultures was higher than the fluorescence intensity from Sec:msfGFP cell cultures, which may reflect differences in the activity of the two different pathways, different rates of msfGFP transcription, translation, or protein folding when fused to a particular signal peptide.
–
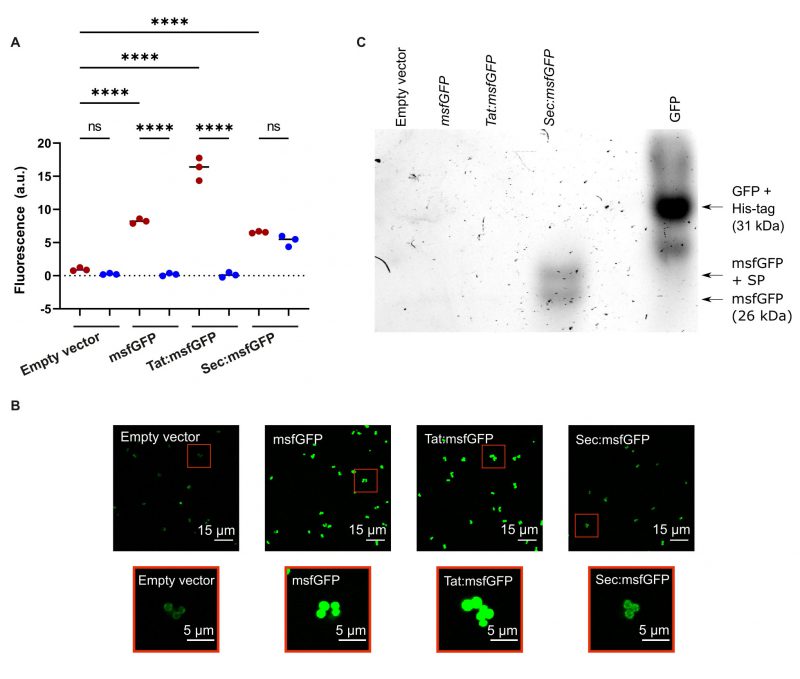 | FIGURE 2. (A) Fluorescence intensity from excitation of msfGFP in cell cultures (red circles) or cell-free supernatants (blue circles) of S. aureus expressing msfGFP from the pRMC2 vector. msfGFP was fused to either Tat or Sec signal peptides, no signal peptide, or not expressed at all (empty vector). Black bars indicate group medians. Samples were compared using a one-way ANOVA followed by a Tukey's test; **** denotes a p < 0.0001 significance level and ns denotes no significance. (B) CLSM images of S. aureus cells expressing msfGFP fusions. Red boxes indicate zoomed in images. All fluorescence images had their brightness increased equally using Fiji ImageJ for clear visualisation. (C) In-gel fluorescence of GFP/msfGFP in a native PAGE gel containing supernatants from cultures expressing the empty pRMC2 vector, msfGFP without a signal peptide, or fused to either a Sec or Tat signal peptide. |
–
The presence of fluorescent msfGFP in the intracellular and extracellular environment was verified by CLSM imaging of cell cultures expressing Tat:msfGFP, Sec:msfFP, msfGFP, and cells containing the empty vector. As expected, msfGFP fluorescence was detected inside cells expressing Tat:msfGFP and msfGFP (Figure 2B). There was a weak fluorescence in S. aureus expressing Sec:msfGFP, which reflects that there was a small fraction of GFP that was not secreted from the bacteria or that remained linked to the cell wall. Furthermore, in-gel fluorescence showed that only the Sec:msfGFP strain secreted a functional msfGFP (Figure 2C). However, the secreted msfGFP was found in two distinct sizes in the supernatant of the Sec:msfGFP cultures, which indicates that the signal peptide was not always removed from some of the msfGFP during secretion. There is an additional band underneath the GFP control (Figure 2C), which is likely GFP that lacks a His-tag. Although we have confirmed msfGFP is found in the supernatant by bulk measurements using a plate reader (Figure 2A), the fluorescence could not be seen in the supernatant using CLSM because the fluorescent protein was too diluted to be visualised. There is also a weak surface-associated fluorescent signal seen with CLSM on S. aureus containing an empty vector without msfGFP, which is due to autofluorescence from ATc [36].
–
Coa:msfGFP produces a functional coagulase that localises within the fibrin pseudocapsule
msfGFP was successfully secreted via the Sec pathway, so to demonstrate its suitability to tag extracellular proteins, it was fused to Coa by insertion into the S. aureus chromosome via allelic exchange. Coa is one of two coagulases that hijack the human coagulation cascade and triggers the formation of a fibrin network around S. aureus cells, a major component of the biofilm extracellular matrix in vivo, that protects S. aureus from the host immune system during infection [29][30]. In order to confirm that the chromosome-integrated coa:msfGFP had not impacted the ability of Coa to cause coagulation, the fusion protein was first created in a mutant strain that lacks the other coagulase: Von Willebrand factor binding protein (vWbp) [37]. Loss of function of coagulase would then result in inability to coagulate plasma.
–
The fusion protein Cos:msfGFP was secreted successfully from S. aureus and the fusion protein did not get cleaved, demonstrated by in-gel fluorescence analysis (Figure 3A). Fluorescence from GFP was present in the supernatant of bacterial cultures expressing Coa:msfGFP and not in cultures without Coa:msfGFP, demonstrating that the fusion protein was secreted extracellularly (Figure 3A). Coa:msfGFP from the supernatant of bacterial cultures did not travel as far through the gel as GFP alone, or msfGFP fused to a Sec signal peptide. Therefore, the weight of the fusion protein was much larger, demonstrating that the protein is intact and contains both Coa and msfGFP (Figure 3A). Coa was also functional, as S. aureus with chromosome-integrated coa:msfGFP coagulated plasma similarly to the parental strains (Figure 3B), and biofilms formed similar fibrin structures as the parental strains, i.e. fibrin was visible as pseudocapsules surrounding clusters of bacteria and as an extended fibrous network between clusters of bacteria (Figure 4A, 4B)). This was true for both the wildtype and the mutant lacking vWbp, thus the fusion to msfGFP did not inhibit the function of Coa. We confirmed that coagulation occurred due to Coa and vWbp alone by including a control mutant of S. aureus that lacks both coa and vwbp, which did not coagulate plasma (Figure 3B) nor produce fibrin fibers in the biofilm matrix (Figure 4C). Fibrin was visualised by the addition of fluorescently labelled fibrinogen to the biofilm growth medium, which is converted into fibrin fibers by an activated complex formed by Coa and vWbp binding to host prothrombin. The small amount of red fluorescence seen in Figure 4C are aggregates of fluorescent fibrinogen that have not been converted into fibrin because of the absence of both Coa and vWbp.
–
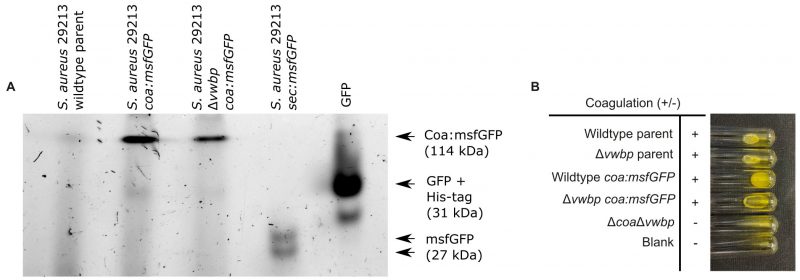 | FIGURE 3: The Coa:msfGFP fusion protein was successfully secreted from S. aureus and functioned correctly. (A) n-gel fluorescence of GFP/msfGFP in a native PAGE gel containing supernatants from the S. aureus wildtype parent strain, as well as S. aureus wildtype and S. aureus Δvwbp both expressing Coa:msfGFP. His-tagged GFP and supernatant from the strain expressing Sec:msfGFP were also loaded to the gel to serve as a molecular marker and as a positive control for GFP and msfGFP fluorescence, respectively. The gel shows that Coa:msfGFP was secreted as an intact, fluorescent protein, giving a band at the expected weight of msfGFP and Coa combined [53]. (B) Coagulation of S. aureus 29213 wildtype and Δvwbp producing either Coa:msfGFP or unmodified Coa and S. aureus 29213 ΔcoaΔvwbp after 24 hours incubation with human plasma at 37°C. All strains producing Coa coagulated plasma, while the double mutant ΔcoaΔvwbp did not. |
–
To assess the location of Coa in S. aureus biofilms, we visualised the bacterial cells, fibrin, and Coa:msfGFP by CLSM. Coa:msfGFP localised to the surface of the bacteria, where we predicted that Coa catalyses the formation of a fibrin pseudocapsule (Figure 4A). This finding corroborates previous studies, which also detected Coa in the fibrin pseudocapsule by immunolabelling [30]. Biofilms of the parental strain were used as negative controls, and here we detected no fluorescence from GFP (Figure 4B). We have thus demonstrated the use of msfGFP for labelling a protein secreted by the Sec pathway in S. aureus. The signal from msfGFP appears brighter in the mutant lacking vWbp, and this is most likely caused by a less dense extracellular matrix in this strain which lacks one of the coagulases, and the signal from msfGFP is therefore attenuated less.
–
 | FIGURE 4. (A) CLSM images of S. aureus wildtype and S. aureus Δvwbp biofilms producing Coa:msfGFP. The composite image (left) is displayed along with the channel containing only signal from Coa:msfGFP (middle) and a zoomed in image of that channel (right). Coa:msfGFP localised to the surface of bacteria within the fibrin pseudocapsule. (B) The parental strains of S. aureus that produce unmodified Coa when imaged with the same imaging settings as modified bacteria producing Coa:msfGFP. No fluorescence was detected, which confirms that the fluorescence in Figure 4A originates from msfGFP and not from autofluorescence. (C) A double mutant lacking both Coa and vWbp (ΔcoaΔvwbp) forms no fibrin at all. Biofilms were grown in BHI containing 50% human plasma for 2 h. Bacteria (wildtype and Δvwbp) were visualised by staining with SYTO 41 (blue), fibrin was visualised by amending Alexa 647-conjugated fibrinogen to media (red), and Coa was visualised by fluorescence emitted from msfGFP (green). The double mutant expressed gfp from a plasmid pCM29 [54] (blue), and fibrin was also visualised by addition of Alexa-647-conjugated fibrinogen to the media (red). Brightness for each colour channel for each image were increased equally using Fiji ImageJ to visualise the data for both (A) and (B). |
DISCUSSION
We show that msfGFP can be used to generate extracellular fluorescent fusion proteins in S. aureus, but that the application is limited to proteins that are secreted through the Sec pathway. When fused to Coa, msfGFP did not hinder the biological function of Coa, and the fusion protein localised to the fibrin pseudocapsule surrounding clusters of S. aureus cells. This result is in agreement with previous studies [30][31] and indicates that fusion to msfGFP does not cause Coa to mislocalise or malfunction. Therefore, msfGFP is a good candidate for tagging S. aureus proteins exported by the Sec pathway, and the majority of extracellular proteins are indeed secreted by this pathway [14].
–
msfGFP has a superfolding mutation that makes it fold quickly and without chaperones, even when fused to another protein, and it exhibits a high level of brightness [34] that makes it ideal for creating fusion proteins in the extracellular environment. Correct folding is essential for chromophore formation and fluorescence, while fast folding is also important for the protein to fold into its 3D conformation in time to avoid cleavage by extracellular proteases that clear unfolded or misfolded proteins away from the cell surface. msfGFP is also monomeric, which makes it less likely to aggregate and cause artefacts, which makes it a good candidate for many fusion proteins. We have demonstrated for the first time the generation of fluorescent fusion proteins for a secreted protein in S. aureus, and this approach now opens possibilities of studying the location of secreted proteins that remain associated with the extracellular matrix of staphylococcal biofilms. The fluorescence signal was fairly dim when imaging Coa:msfGFP, however, it is not known how much Coa is produced and therefore the concentration could be low. Additionally, we imaged the fusion protein in the complex environment of a biofilm. Biofilms are thick, heterogeneous samples that are autofluorescent and attenuate and distort both the excitation and emission from fluorescent molecules, which makes them a challenging environment to image in. However, the fact that Coa:msfGFP could be visualised by standard CLSM imaging is encouraging, and advanced microscopes with more sensitive detection will facilitate more detailed analyses. For example, single-molecule microscopy on live bacteria has revealed important details of Tat-mediated transport in Gram-negative E. coli, and similar investigations could be pursued for Sec-secreted proteins in Gram-positive bacteria using msfGFP fusions [21]. In particular, future studies could utilise total internal reflection fluorescence (TIRF) microscopy to investigate extracellular secretion between the cell and a surface such as an agarose pad or glass coverslip, that are sensitive at single-molecule GFP detection limits in live bacteria [38][39], or faster millisecond Slimfield microscopy that could potentially enable mobility studies of extracellular secreted components [40][41][42].
–
S. aureus is a major biofilm-forming human pathogen that establishes infections, causes disease, and evades the immune system through a number of secreted and cell surface associated proteins, many of which contain a Sec-type signal peptide [23]. Fusions with msfGFP will greatly benefit future research into these proteins. We do not know whether msfGFP would be exported correctly via the Sec pathway in other bacterial species; past studies into GFP export via the Tat pathway in Gram positive bacteria revealed that a different GFP variant was not exported correctly in all species tested [6]. The authors speculated that their results were due to differences in the physical or chemical structure of the cell wall, or in the quality control mechanisms of the Tat translocases. Such interspecies differences may also affect the outcome when using msfGFP for Sec exported proteins, and this is important to bear in mind. The assay performed by expression of msfGFP from pRMC2 vector in our study, however, provides an easy tool for checking the feasibility of msfGFP secretion in other Staphylococci that are compatible with this vector, and the Tat and Sec signal peptide constructs can be cloned into different vectors for studies in other Gram-positive bacteria.
–
We have confirmed that msfGFP is a good candidate for labelling proteins secreted by the Sec pathway in S. aureus. We fused msfGFP to coa in the S. aureus chromosome and demonstrated that fusion to msfGFP did not prevent Coa from functioning correctly, that msfGFP could fold correctly and fluoresce in the extracellular environment, and that the fusion protein localised as expected in the extracellular environment. S. aureus utilises a myriad of surface associated and secreted proteins to establish infections and cause disease, and our work opens the door for developing fusion proteins to investigate these and progress our understanding of S. aureus infection.
MATERIALS AND METHODS
Materials, bacterial strains, and growth conditions
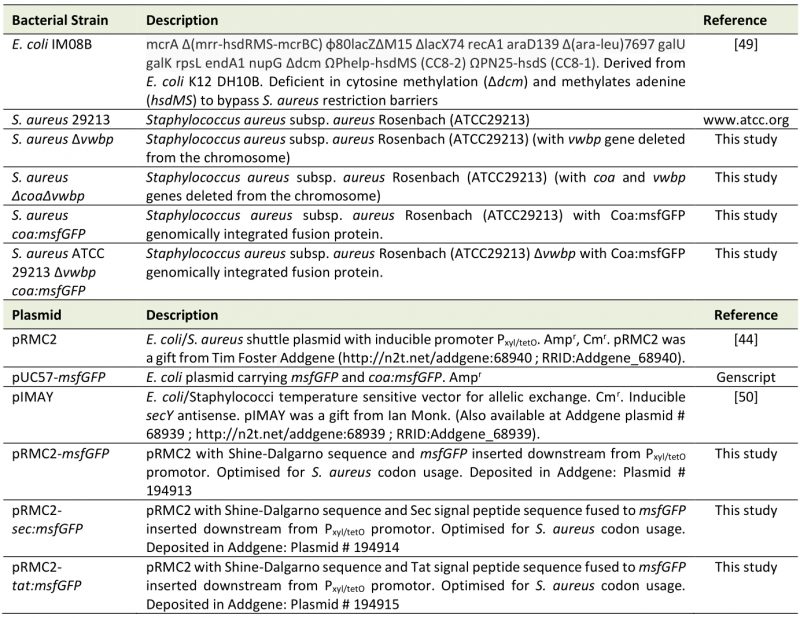
All bacterial strains, plasmids, and primers used are listed in Table 1. For long-term storage, bacteria were stored in 25% glycerol at -80°C. E. coli and S. aureus were cultured in Luria Broth (LB, L3522, Sigma-Aldrich) and Brain Heart Infusion (BHI, 53286, Millipore), respectively, at 37°C with 180 rpm shaking. When grown on agar, 15 g/L agar (A1296, Sigma-Aldrich) was added to the media. For plasmid selection, the media was supplemented with 25 µg/ml or 10 µg/ml chloramphenicol (Cm, C0378, Sigma-Aldrich), or 100 µg/ml ampicillin (Amp, A9393, Sigma-Aldrich). Biofilms were grown in modified BHI (mBHI) supplemented with 50% heparin stabilised human plasma to mimic physiological conditions. mBHI is BHI supplemented with 2.1 mM CaCl2 (C3881, Sigma-Aldrich) and 0.4 mM MgCl2 (31413, Sigma-Aldrich). When low autofluorescence conditions were required, bacteria were suspended in mM9 medium. mM9 is a minimal medium comprising of M9 salts (M6030, Sigma-Aldrich) supplemented with 2 mM MgSO4 (M1880, Sigma-Aldrich), 0.1 mM CaCl2 (C3881, Sigma-Aldrich), 1% glucose (1.08346, Merck), 1% casamino acids (Gibco, 223050), 1 mM Thiamine-HCl (T4625, Sigma-Aldrich), and 0.05 mM nicotinamide (72340, Sigma-Aldrich) [43]. Plasma was collected from blood donated by Aarhus University Hospital by centrifugation at 2000 x g for 15 minutes at 4°C and stored in aliquots at -80 °C. Before use, frozen plasma was immediately thawed in a water bath at 37°C. Tat and Sec signal peptide fusion protein expression was induced with the addition of 340 ng/ml anhydrotetracycline (ATc, 94664, Sigma-Aldrich).
–
Table 1. Bacterial strains and plasmids used in this study. |
 |
–
Construction of pRMC2 overexpression vector carrying signal peptide:msfGFP constructs
Tat and Sec signal peptide sequences were fused to msfGFP to create tat:msfGFP and sec:msfGFP in the vector pRMC2 (Figure 1), a plasmid with an inducible Pxyl/tetO promoter and origin of replication for E. coli and Staphylococci (Table 1) [44]. A positive control was also constructed expressing msfGFP with no signal peptide (Figure 1). Note that the Shine-Dalgarno sequences were added later as described in the following section.
–
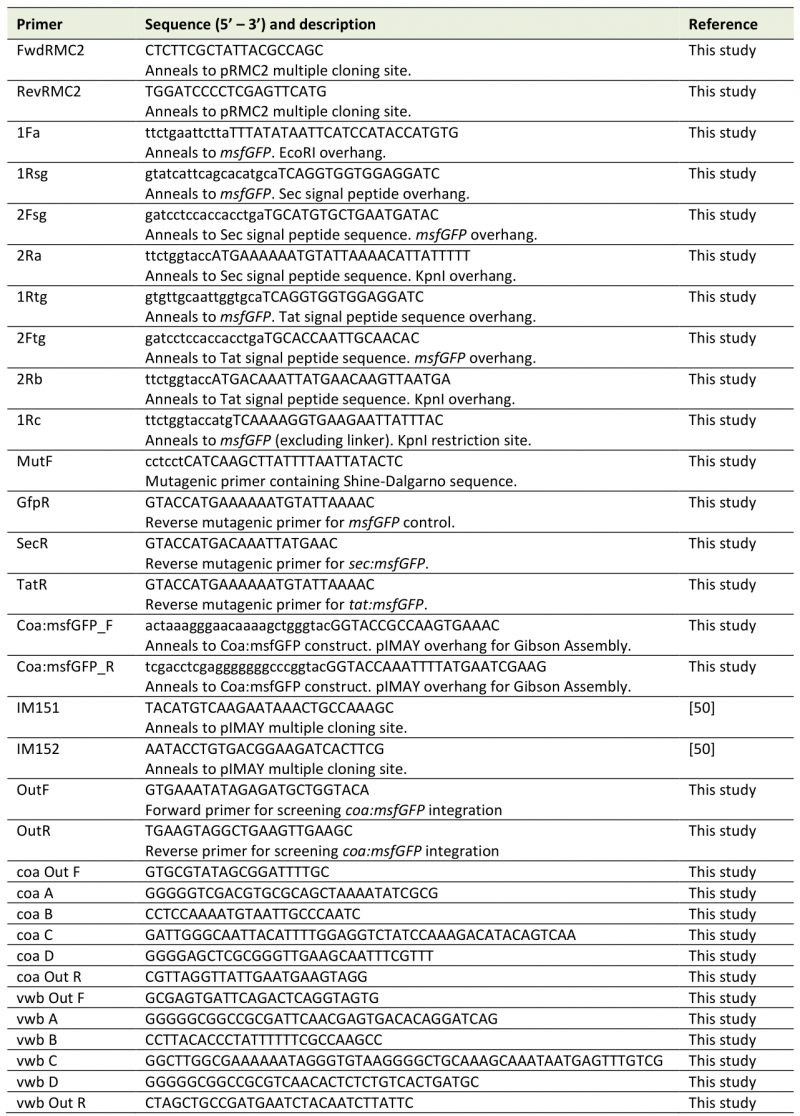
Sequences for msfGFP [34], Tat [18], and Sec signal peptides [45] were reverse translated with an S. aureus USA300 codon usage table (see Supplementary Table S1 for sequences). The RNA polymerase α and β subunits are highly conserved, and their nucleotide sequences were used to predict codon usage in S. aureus USA300 and S. aureus 29213, and an S. aureus USA300 codon usage table was deemed suitable. Signal peptide sequences were ordered as oligos (Thermo Fisher Scientific) and msfGFP with a linker at its N-terminal was ordered on a high copy plasmid (pUC57, Genscript). The signal peptide sequences and msfGFP were amplified by PCR with Phusion polymerase (F566S, Thermo Fisher Scientific) according to the manufacturer's instructions. The primers (Invitrogen), listed in Table 2, contained overhangs intended to join fragments and add KpnI and EcoRI restriction sites at the 5' and 3' ends. Primers 2Ftg/2Rb and 2Fsg/2Ra were used to amplify Tat and Sec signal peptide sequences, respectively, and msfGFP was amplified with 1Fa/1Rsg. The signal peptide sequences were joined to msfGFP via SOE-PCR to create tat:msfGFP (primers 1Fa/Rtg) and sec:msfGFP (primers 1Fa/2Ra). msfGFP was also amplified alone with no signal peptide sequence to be used as a control. PCR products were analysed by gel electrophoresis and purified with the GenElute Gel Extraction Kit (NA1111, Sigma-Aldrich). All PCR products and pRMC2 were digested by KpnI (FD0524, Thermo Fisher Scientific) and EcoRI (FD0274, Thermo Fisher Scientific), and PCR products were ligated into pRMC2 with T4 DNA ligase (EL0011, Invitrogen) according to the manufacturer's protocols.
–
Table 2. Primers used in this study. Annealing sequence of primers is given in upper case, and overhangs in lower case text. |
 |
–
Insertion of Shine-Dalgarno sequence via site directed mutagenesis
In order to make the translation of msfGFP possible, the Shine-Dalgarno sequence was inserted upstream of the signal peptide and msfGFP sequences via site directed mutagenesis [46]. The consensus sequence was chosen [47] and inserted 5 nucleotides upstream of the start codons of tat:msfGFP, sec:msfGFP, and msfGFP to ensure maximum translation efficiency [48]. To do this, a mutagenic primer, MutF, was designed with an overhang containing the Shine-Dalgarno sequence and used to amplify the entire pRMC2 constructs containing tat:msfGFP, sec:msfGFP and msfGFP and simultaneously insert the sequence at the desired place. A unique reverse primer was designed for each construct, while the mutagenic primer MutF remained the same (MutF/TatR for tat:msfGFP, MutF/SecR for sec:msfGFP, and MutF/GfpR for msfGFP). The primers were phosphorylated using T4 Polynucleotide Kinase (EK0031, Thermo Fisher Scientific) according to the manufacturer's instructions. The constructs were then amplified using the phosphorylated primers and Phusion polymerase (Phusion Hot Start II DNA Polymerase, F549S, Thermo Fisher Scientific) according to the manufacturer's instructions. The new PCR products were digested with DpnI to remove methylated template DNA, after which the mutated plasmids were ligated back into a whole plasmid according to the manufacturer's instructions (Phusion Site-Directed Mutagenesis Kit, F541, Thermo Fisher Scientific).
–
Transformation into E. coli IM08B
The pRMC2 constructs expressing Tat:msfGFP, Sec:msfGFP, or msfGFP, and empty pRMC2, were first transformed via heat shock into E. coli IM08B in order to gain a methylation profile mimicking S. aureus [49]. To prepare chemical competent cells, an overnight culture of E. coli IM08B was diluted to OD600 0.02 and grown to OD600 0.3, then chilled on ice for 10 minutes. Cells were harvested by centrifugation at 4000 x g for 10 minutes at 4°C and resuspended in 5 ml ice cold 0.5 M CaCl2. The centrifugation was repeated, and the cells resuspended in 1.2 ml 0.5 M CaCl2 before incubating on ice for 30 minutes. For transformation, 1-3 µl of each pRMC2 construct was incubated for 30 minutes on ice with 50 µl of competent cells. A heat shock was applied at 4 °C for 90 s, and cells were then transferred to ice for 2 minutes. 950 µl of preheated LB media (37°C) was added and then cells incubated with 180 rpm shaking for 1 hour at 37°C. Cells were finally plated on agar with Amp and incubated at 37°C overnight. Plasmids were extracted from positive transformants with the GeneJET Plasmid Miniprep Kit (K0502, Sigma-Aldrich) and sent for sequencing with Macrogen Europe with primers FwdRMC2/RevRMC2.
–
Transformation into S. aureus 29213
Plasmids with the correct sequence were transformed into S. aureus 29213 by electroporation. To prepare electrocompetent cells, an overnight culture was diluted to OD600 0.5 and grown to OD600 0.6. Cells were harvested by centrifugation at 4000 x g for 10 minutes at 4°C and washed in 50 ml ice cold MilliQ water three times. Cells were then centrifuged and resuspended in 50 ml, then 5 ml, 2 ml, and finally 0.25 ml ice cold 0.5 M sucrose. Up to 1 µg plasmid DNA was incubated on ice with 50 µl fresh competent cells for 10 minutes before being transferred to a chilled 1 mm electroporation cuvette and electroporated at 2.1 kV, 200 Ω, and 25 µF in an ECM 630 BTXTM (Harvard Apparatus). Immediately afterwards, 1 ml preheated BHI supplemented with 0.5 M sucrose (37°C) was added to the cells, which were then incubated at 37°C with 150 rpm shaking for 2 hours. Cells were finally plated on agar containing Cm and incubated overnight at 37°C. Positive transformants were confirmed by sequencing as described in the prior section.
–
Creation of gene deletion mutants
In-frame single deletions of the coa and vwbp genes were achieved through splicing by overlap extension PCR according to Monk and colleagues [50] and performed as described in detail in Wassmann et al. 2022 [51]. The double mutant was created by introducing the pIMAYΔcoa plasmid into the Δvwbp mutant and deleting the coa gene in the Δvwbp mutant.
–
Construction and evaluation of a chromosome-integrated Coa:msfGFP fusion protein
A C-terminal, chromosome-integrated fusion Coa:msfGFP was created by allelic replacement using the protocol from Monk et al. [50]. Primers Coa:msfGFP_F/Coa:msfGFP_R were used to amplify coa:msfGFP from a pUC57-msfgfp and add overhangs for Gibson Assembly. pIMAY was digested using restriction enzyme KpnI (R3142S, New England Biolabs) and then ligated to coa:msfGFP via Gibson Assembly [52] using a kit (E5510S, New England Biolabs). The ligated construct was first transformed via chemical transformation into E. coli IM08B to gain a methylation profile mimicking that of S. aureus [49], and was then extracted and transformed via electroporation into S. aureus 29213 wildtype and Δvwbp, a mutant lacking vWbp, the other coagulase that S. aureus produces [37]. After transformation into S. aureus, the plasmid was then integrated into the chromosome and finally the backbone was excised using the protocol from Monk et al. [50]. The genotype of the fusion protein was assessed via sequencing with the OutR/OutF primers and the phenotype was assessed via coagulation assays. For coagulation assays, overnight cultures of the mutant and parental strains were diluted to OD600 0.5 in 1 ml of 1:6 heparin-stabilised human plasma in 0.8 % NaCl (w/v) (S5886, Sigma-Aldrich) in sterile glass tubes and incubated for 24 hours at 37°C with no shaking. Coagulation was assessed by tilting and observing the tubes after 24 hours incubation. A negative control without bacteria was also included in addition to the mutant lacking both Coa and vWbp, which should not coagulate plasma.
–
Screening for msfGFP fluorescence in cell cultures and supernatants by bulk fluorescence
To verify if msfGFP was successfully secreted by the Tat and Sec pathways, the supernatants of bacteria expressing the signal peptide and msfGFP fusions were investigated for fluorescence by bulk and in-gel measurements. Bacterial cultures and supernatants from S. aureus 29213 expressing Tat:msfGFP, Sec:msfGFP, msfGFP, or no msfGFP from the overexpression vector pRMC2 were grown overnight in BHI and diluted to OD600 0.1 in mM9 medium and incubated at 37 °C with 180 rpm shaking until OD600 0.5. mM9 was used in place of BHI because it is less autofluorescent. ATc (340 ng/ml) was added to the cultures, after which they were further incubated for 60 minutes to induce the Pxyl/tetO promoter and msfGFP expression. Final OD600 was recorded, and 2 ml of each sample taken. For the fluorescence bulk measurements, 200 µl was added directly into a 96-well plate (Nunc F96 MicroWell Black-bottom plate, 237105, Thermo Fisher Scientific) and the remaining 1.8 ml was centrifuged at 14104 x g for 10 minutes. The supernatant was removed, sterile filtered with a 0.2 µm filter (83.1826.001, Sarstedt), and 200 µl was added to the 96-well plate. Three biological replicates (from independently grown cultures) and three technical replicates (individually prepared samples from the same culture) were tested per construct. Fluorescence was measured at 488 nm wavelength excitation, 510 nm wavelength emission, and 1000 ms exposure time in a Varioskan Lux Flash Plate Reader (Thermo Fisher Scientific). Median fluorescence values were calculated and normalised to the optical density. The values were tested for normality with a Shapiro-Wilk test, after which they were compared using a one-way ANOVA followed by a Tukey's test with a p < 0.05 significance level.
–
Visualisation of secreted msfGFP and Coa:msfGFP via in-gel fluorescence
To verify whether Sec:msfGFP and Tat:msfGFP were secreted and folded correctly, and to confirm whether Coa:msfGFP was secreted as an intact, functionally fluorescent protein, we separated the supernatant on a native PAGE gel. Cultures that expressed either Coa:msfGFP, Tat:msfGFP, msfGFP, an empty vector were grown to the exponential phase in mM9, and the chromosome-integrated Coa:msfGFP cultures were grown to the stationary phase in BHI, after which the supernatant was collected and either stored at –80°C or used right away. The supernatants were mixed 1:1 with a native sample buffer (1610738, Bio-Rad) and separated on a 4–15% precast polyacrylamide protein gel (Mini-PROTEAN TGX, 4561086, Bio-Rad). Fluorescence was detected in the Amersham Typhoon Scanner (29187191, Cytiva) with a 488 nm excitation and 510 nm emission. A His-tagged GFP (14-392, Sigma-Aldrich) was also loaded on the gel and used as a positive control for GFP fluorescence as well as a size marker.
–
Confocal microscopy of S. aureus expressing signal peptide fusions
To visualise whether msfGFP was retained within cells, all overexpression constructs were also imaged with confocal laser scanning microscopy (CLSM). Overnight cultures were diluted to OD600 0.1 in mM9 medium and were grown to OD600 0.5, then incubated for a further 2 hours with 340 ng/ml ATc and imaged with the LSM700 confocal microscope (Zeiss) with a 10 mW 488nm wavelength laser at 2% power and a Plan-Apochromat 63x/1.40 NA oil immersion objective lens. Images were captured with the Axiocam HR camera (Zeiss) and using the Zen Black software (Zeiss).
–
Confocal microscopy of S. aureus expressing Coa:msfGFP
S. aureus expressing either the chromosome-integrated fusion Coa:msfGFP or unmodified Coa were grown overnight in BHI and then diluted to OD600 5. Microwells (µ-Slide 8 Well, 80821, IBIDI) were preconditioned with 180 µl BHI supplemented with 50% plasma, 10 µM Syto41 (S11352, Invitrogen), and 0.4 µg/ml Alexa Fluor 647-conjugated fibrinogen (F35200, Invitrogen) by incubating at 37°C for 30 minutes. Then 20 µl OD600 5 cultures were added and incubated for a further 2 hours. The biofilms were imaged with 405 nm, 488 nm, and 639 nm wavelength excitation and a Plan-Apochromat 63x/1.40 NA oil immersion objective in the LSM700 confocal microscope (Zeiss). Images were captured with the Axiocam HR camera (Zeiss) and using the Zen Black software (Zeiss). GFP fluorescence was detected with 488 nm wavelength excitation and 490-600 nm wavelength emission, and Alexa 647-conjugated fibrinogen was detected with 639 nm wavelength excitation and 640-750 nm emission.
REFERENCES
- Chiu SW and Leake MC (2011). Functioning nanomachines seen in real-time in living bacteria using single-molecule and super-resolution fluorescence imaging. Int J Mol Sci 12(4): 2518–2542. 10.3390/ijms12042518
- Chudakov DM, Matz MV, Lukyanov S, and Lukyanov KA (2010). Fluorescent proteins and their applications in imaging living cells and tissues. Physiol Rev 90(3): 1103–1163. 10.1152/physrev.00038.2009
- Linton E, Walsh MK, Sims RC, and Miller CD (2012). Translocation of green fluorescent protein by comparative analysis with multiple signal peptides. Biotechnol J 7(5): 667–676. 10.1002/biot.201100158
- Thomas JD, Daniel RA, Errington J, and Robinson C (2001). Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol 39(1): 47–53. 10.1046/j.1365-2958.2001.02253.x
- Li SY, Chang BY, and Lin SC (2006). Coexpression of TorD enhances the transport of GFP via the TAT pathway. J Biotechnol 122(4): 412–421. 10.1016/j.jbiotec.2005.09.011
- Meissner D, Vollstedt A, Van Dijl JM, and Freudl R (2007). Comparative analysis of twin-arginine (Tat)-dependent protein secretion of a heterologous model protein (GFP) in three different Gram-positive bacteria. Appl Microbiol Biotechnol 76(3): 633–642. 10.1007/s00253-007-0934-8
- van der Ploeg R, Monteferrante CG, Piersma S, Barnett JP, Kouwen TR, Robinson C, and van Dijl JM (2012). High-salinity growth conditions promote tat-independent secretion of tat substrates in Bacillus subtilis. Appl Environ Microbiol 78(21): 7733–7744. 10.1128/AEM.02093-12
- Snyder AJ, Mukherjee S, Glass JK, Kearns DB, and Mukhopadhyay S (2014). The canonical twin-arginine translocase components are not required for secretion of folded green fluorescent protein from the ancestral strain of Bacillus subtilis. Appl Environ Microbiol 80(10): 3219–3232. 10.1128/AEM.00335-14
- Guo L, Wu T, Hu W, He X, Sharma S, Webster P, Gimzewski JK, Zhou X, Lux R, and Shi W (2013). Phenotypic characterization of the foldase homologue PrsA in Streptococcus mutans. Mol Oral Microbiol 28(2): 154–165. 10.1111/omi.12014
- Cowley SC and Av-Gay Y (2001). Monitoring promoter activity and protein localization in Mycobacterium spp. using green fluorescent protein. Gene 264(2): 225–231. 10.1016/S0378-1119(01)00336-5
- Hoffmann A, Schneider T, Pag U, and Sahl HG (2004). Localization and functional analysis of PepI, the immunity peptide of Pep5-producing Staphylococcus epidermidis strain 5. Appl Environ Microbiol 70(6): 3263–3271. 10.1128/AEM.70.6.3263-3271.2004
- Knapp A, Ripphahn M, Volkenborn K, Skoczinski P, and Jaeger KE (2017). Activity-independent screening of secreted proteins using split GFP. J Biotechnol 258: 110–116. 10.1016/j.jbiotec.2017.05.024
- Wiedenmann J, Oswald F, and Nienhaus GU (2009). Fluorescent proteins for live cell imaging: Opportunities, limitations, and challenges. IUBMB Life 61(11): 1029–1042. 10.1002/iub.256
- Prabudiansyah I, Driessen AJM (2017). The Canonical and Accessory Sec System of Gram-positive Bacteria. Curr Top Microbiol Immunol 404: 45-67. 10.1007/82_2016_9
- Green ER and Mecsas J (2016). Bacterial Secretion Systems: An Overview. Microbiol Spectr 4(1): 1–19. 10.1128/microbiolspec.vmbf-0012-2015
- Papanikou E, Karamanou S, and Economou A (2007). Bacterial protein secretion through the translocase nanomachine. Nat Rev Microbiol 5(11): 839–851. 10.1038/nrmicro1771
- Robinson C and Bolhuis A (2004). Tat-dependent protein targeting in prokaryotes and chloroplasts. Biochim Biophys Acta – Mol Cell Res 1694(1-3): 135–147. 10.1016/j.bbamcr.2004.03.010
- Biswas L, Biswas R, Nerz C, Ohlsen K, Schlag M, Schäfer T, Lamkemeyer T, Ziebandt AK, Hantke K, Rosenstein R, Götz F (2009). Role of the twin-arginine translocation pathway in Staphylococcus. J Bacteriol 191(19): 5921–5929. 10.1128/JB.00642-09
- Müller M (2005). Twin-arginine-specific protein export in Escherichia coli. Res Microbiol 156(2): 131–136. 10.1016/j.resmic.2004.09.016
- Berks BC, Palmer T, and Sargent F (2005). Protein targeting by the bacterial twin-arginine translocation (Tat) pathway. Curr Opin Microbiol 8(2): 174–181. 10.1016/j.mib.2005.02.010
- Leake MC, Greene NP, Godun RM, Granjon T, Buchanan G, Chen S, Berry RM, Palmer T, and Berks BC (2008). Variable stoichiometry of the TatA component of the twin-arginine protein transport system observed by in vivo single-molecule imaging. Proc Natl Acad Sci USA 105(40): 15376–15381. 10.1073/pnas.0806338105
- Tong SYC, Davis JS, Eichenberger E, Holland TL, and Fowler VG (2015). Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28(3): 603–661. 10.1128/CMR.00134-14
- Foster TJ, Geoghegan JA, Ganesh VK, and Höök M (2014). Adhesion, invasion and evasion: The many functions of the surface proteins of Staphylococcus aureus. Nat Rev Microbiol 12(1): 49–62. 10.1038/nrmicro3161
- Crosby HA, Kwiecinski J, Horswill AR, Roy J, and City I (2016). Staphylococcus aureus aggregation and coagulation mechanisms, and their function in host-pathogen interactions. Adv Appl Microbiol 96: 1–41. 10.1016/bs.aambs.2016.07.018.Staphylococcus
- Stapels DA, Ramyar KX, Bischoff M, von Köckritz-Blickwede M, Milder FJ, Ruyken M, Eisenbeis J, McWhorter WJ, Herrmann M, van Kessel KP, Geisbrecht BV, and Rooijakkers SH (2014). Staphylococcus aureus secretes a unique class of neutrophil serine protease inhibitors. Proc Natl Acad Sci USA 111(36): 13187–13192. 10.1073/pnas.1407616111
- Hussain M, Becker K, Von Eiff C, Schrenzel J, Peters G, and Herrmann M (2001). Identification and characterization of a novel 38.5-Kilodalton cell surface protein of Staphylococcus aureus with extended-spectrum binding activity for extracellular matrix and plasma proteins. J Bacteriol 183(23): 6778–6786. 10.1128/JB.183.23.6778-6786.2001
- Ko YP, Kuipers A, Freitag CM, Jongerius I, Medina E, van Rooijen WJ, Spaan AN, van Kessel KP, Höök M, Rooijakkers SH (2013). Phagocytosis Escape by a Staphylococcus aureus Protein That Connects Complement and Coagulation Proteins at the Bacterial Surface. PLoS Pathog 9(12): 1–13. 10.1371/journal.ppat.1003816
- Palma M, Nozohoor S, Schennings T, Heimdahl A, and Flock JI (1996). Lack of the extracellular 19-kilodalton fibrinogen-binding protein from Staphylococcus aureus decreases virulence in experimental wound infection. Infect Immun 64(12): 5284–5289. 10.1128/iai.64.12.5284-5289.1996
- McAdow M, Missiakas DM, and Schneewind O (2012). Staphylococcus aureus secretes coagulase and von willebrand factor binding protein to modify the coagulation cascade and establish host infections. J Innate Immun 4(2): 141–148. 10.1159/000333447
- Guggenberger C, Wolz C, Morrissey JA, and Heesemann J (2012). Two distinct coagulase-dependent barriers protect Staphylococcus aureus from neutrophils in a three dimensional in vitro infection model. PLoS Pathog 8(1). 10.1371/journal.ppat.1002434
- Cheng AG, Mcadow M, Kim HK, Bae T, and Missiakas DM (2010). Contribution of Coagulases towards Staphylococcus aureus Disease and Protective Immunity. PLoS Pathog 6(8): e1001036. 10.1371/journal.ppat.1001036
- Vanassche T, Verhaegen J, Peetermans WE, VAN Ryn J, Cheng A, Schneewind O, Hoylaerts MF, Verhamme P (2011). Inhibition of staphylothrombin by dabigatran reduces Staphylococcus aureus virulence. J Thromb Haemost 9: 2436–2446. 10.1111/j.1538-7836.2011.04529.x
- Reth M (2013). Matching cellular dimensions with molecular sizes. Nature Immunol 14: 765-767. 10.1038/ni.2621
- Ke N, Landgraf D, Paulsson J, and Berkmen M (2016). Visualization of periplasmic and cytoplasmic proteins with a self-labeling protein tag. J Bacteriol 198(7): 1035–1043. 10.1128/JB.00864-15
- Sibbald MJ, Ziebandt AK, Engelmann S, Hecker M, de Jong A, Harmsen HJ, Raangs GC, Stokroos I, Arends JP, Dubois JY, van Dijl JM (2006). Mapping the Pathways to Staphylococcal Pathogenesis by Comparative Secretomics. Microbiol Mol Biol Rev 70(3): 755–788. 10.1128/mmbr.00008-06
- Scholz O, Schubert P, Kintrup M, and Hillen W (2000). Tet repressor induction without Mg2+. Biochemistry 39(35): 10914–10920. 10.1021/bi001018p
- Bjerketorp J, Nilsson M, Ljungh Å, Flock JI, Jacobsson K, and Frykberg L (2002). A novel von Willebrand factor binding protein expressed by Staphylococcus aureus. Microbiology 148: 2037–2044. 10.1099/00221287-148-7-2037
- Leake MC, Chandler JH, Wadhams GH, Bai F, Berry RM, and Armitage JP (2006). Stoichiometry and turnover in single, functioning membrane protein complexes. Nature 443(7109): 355–358. 10.1038/nature05135
- Delalez NJ, Wadhams GH, Rosser G, Xue Q, Brown MT, Dobbie IM, Berry RM, Leake MC, and Armitage JP (2010). Signal-dependent turnover of the bacterial flagellar switch protein FliM. Proc Natl Acad Sci USA 107(25): 11347–11351. 10.1073/pnas.1000284107
- Plank M, Wadhams GH, and Leake MC (2009). Millisecond timescale slimfield imaging and automated quantification of single fluorescent protein molecules for use in probing complex biological processes. Integr Biol 1(10): 602–612. 10.1039/b907837a
- Reyes-Lamothe R, Sherratt DJ, and Leake MC (2010). Stoichiometry and architecture of active DNA replication machinery in Escherichia coli. Science 328(5977): 498–501. 10.1126/science.1185757
- Jin X, Lee JE, Schaefer C, Luo X, Wollman AJM, Payne-Dwyer AL, Tian T, Zhang X, Chen X, Li Y, McLeish TCB, Leake MC, and Bai F (2021). Membraneless organelles formed by liquid-liquid phase separation increase bacterial fitness. Sci Adv 7(43): eabh2929. 10.1126/sciadv.abh2929
- Reed P, Atilano ML, Alves R, Hoiczyk E, Sher X, Reichmann NT, Pereira PM, Roemer T, Filipe SR, Pereira-Leal JB, Ligoxygakis P, and Pinho MG (2015). Staphylococcus aureus Survives with a Minimal Peptidoglycan Synthesis Machine but Sacrifices Virulence and Antibiotic Resistance. PLoS Pathog 11(5): 1–19. 10.1371/journal.ppat.1004891
- Corrigan RM and Foster TJ (2009). An improved tetracycline-inducible expression vector for Staphylococcus aureus. Plasmid 61(2): 126–129. 10.1016/j.plasmid.2008.10.001
- Schallenberger MA, Niessen S, Shao C, Fowler BJ, and Romesberg FE (2012). Type I signal peptidase and protein secretion in Staphylococcus aureus. J Bacteriol 194(10): 2677–2686. 10.1128/JB.00064-12
- Kunkel TA (1985). Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci USA 82(2): 488–492. 10.1073/pnas.82.2.488
- Vimberg V, Tats A, Remm M, and Tenson T (2007). Translation initiation region sequence preferences in Escherichia coli. BMC Mol Biol 8: 1–13. 10.1186/1471-2199-8-100
- McLaughlin JR, Murray CL, and Rabinowitz JC (1981). Unique features in the ribosome binding site sequence of the gram-positive Staphylococcus aureus β-lactamase gene. J Biol Chem 256(21): 11283–11291. 10.1016/S0021-9258(19)68589-3
- Monk IR, Tree JJ, Howden BP, Stinear TP, and Foster TJ (2015). Complete bypass of restriction systems for major staphylococcus aureus lineages. MBio 6(3): 1–12. 10.1128/mBio.00308-15
- Monk IR, Shah IM, Xu M, Tan MW, and Foster TJ (2012). Transforming the untransformable: Application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio 3(2): 1–11. 10.1128/mBio.00277-11
- Wassmann CS, Rolsted AP, Lyngsie MC, Torres-Puig S, Kronborg T, Vestergaard M, Ingmer H, Pontoppidan SP, Klitgaard JK (2022). The menaquinone pathway is important for susceptibility of Staphylococcus aureus to the antibiotic adjuvant, cannabidiol. Microbiol Res 257. 10.1016/j.micres.2022.126974
- Gibson DG, Young L, Chuang RY, Venter JC, Hutchison CA, and Smith HO (2009). Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6(5): 343–345. 10.1038/nmeth.1318
- Boden MK and Flock JI (1989). Fibrinogen-binding protein/clumping factor from Staphylococcus aureus. Infec Immun 57(8): 2358-2363. 10.1128/iai.57.8.2358-2363.1989
- Qazi SNA, Rees CED, Mellits KH, and Hill PJ (2001). Development of gfp vectors for expression in Listeria monocytogenes and other low G+C gram positive bacteria. Microb Ecol 41(4): 301–309. 10.1007/s002480000091
–
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
ML was supported by the BBSRC (BB/R001235/1), EPSRC (EP/T002166/1), the Leverhulme Trust (RPG-2017-340), and the Carlsberg Founda-tion (CF16-0342). We thank Melissa Eriksen for assisting in primer design.
COPYRIGHT
© 2023

GFP fusions of Sec-routed extracellular proteins in Staphylococcus aureus reveal surface-associated coagulase in biofilms by Evans et al. is licensed under a Creative Commons Attribution 4.0 International License.








