
Back to article: Exacerbating and reversing lysosomal storage diseases: from yeast to humans
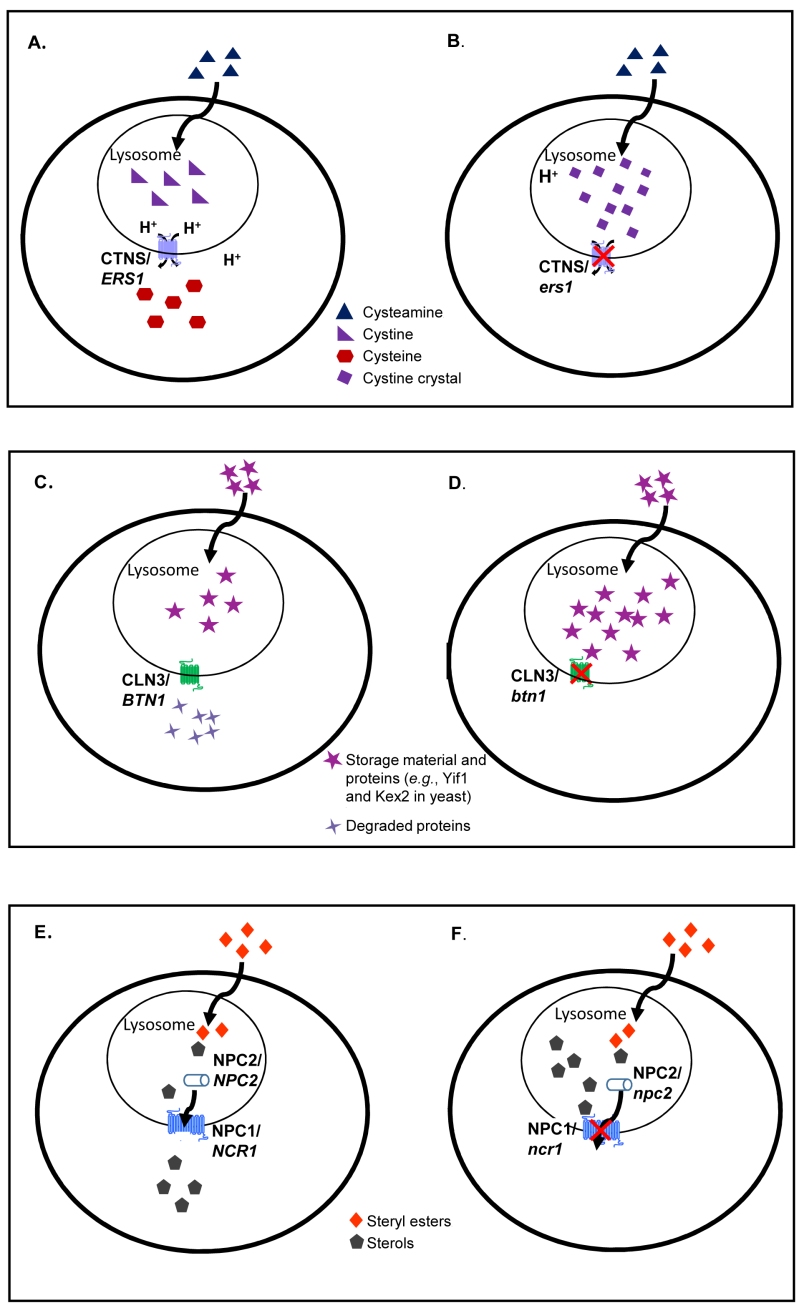
FIGURE 2: Comparisons of normal and mutant cells for three LSDs caused by lysosomal membrane protein deficiencies. In normal cells (A), cystine is exported out of lysosomes to form cysteine. In cystinosis (B), mutations in the disease-causing gene (CTNS, an ortholog of yeast ERS1) prevent the efflux of cystine out of lysosomes causing the formation of cystine crystal in the absence of functional cystinosin. In normal cells (C), autofluorescent storage material and proteins are degraded and transported out of lysosomes. In Batten disease (D), mutations in the disease-causing gene (CLN3, an ortholog of yeast BTN1) limit protein degradation causing an accumulation in the lysosome. In normal cells (E), steryl esters are internalized by receptor mediated endocytosis, undergo lipolysis in the lysosome and egress from lysosomes as free sterols. In NP-C disease (F), mutations in NPC1 (an ortholog of yeast NCR1) prevent the egress of sterol from the lysosome. The same disease phenotype is seen with NPC2 (the human ortholog of yeast NPC2) mutations. In an astounding example of evolutionary conservation, the yeast and human orthologs appear to be interchangeable for all three diseases.