
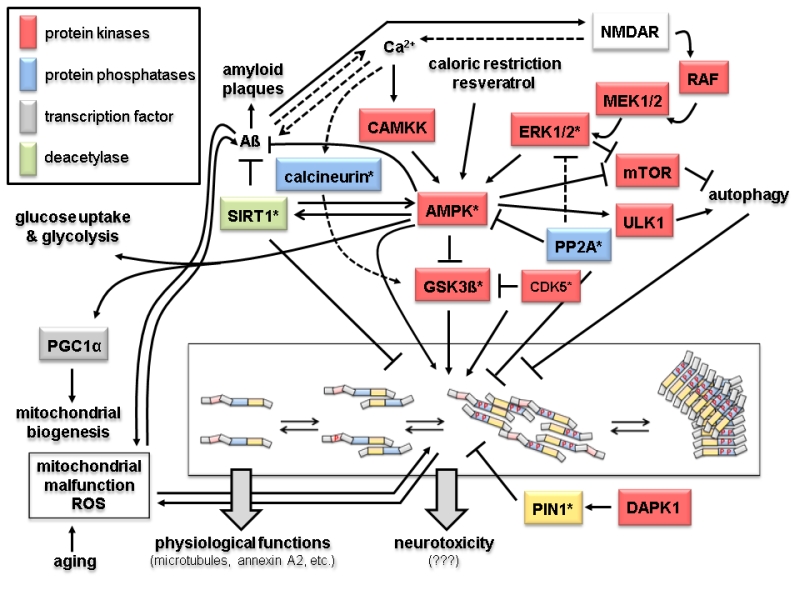
FIGURE 2: Signaling and modification pathways influencing the intracellular concentration of the toxic oligomeric forms of tau. Arrows indicate activation of enzymes or promotion of a conformational change, lines with bars indicate their inhibition/inactivation. Different protein functions are colour-coded as shown in the upper left hand corner. Schematic representation of tau oligomerization states (central lower part) is as in Fig. 1B. Enzymes with homologs in yeast discussed in the text are marked by an asterisk. Question marks with the neurotoxicity output indicate that the exact molecular mechanisms leading to that condition are not known. Despite the number of different protein functions and signaling cascades displayed in this scheme, not all possible components and interacting networks influencing tau oligomerization are shown. Neither are all possible crosstalks between the depicted components shown (e.g. the protein phosphatase PP2A will also inactivate the upstream components of the ERK1/2 MAPK cascade, calcineurin also acts directly on phosphorylated tau, and PIN1 regulates the activities of GSK3ß and AMPK). In the following, the different signaling pathways which affect tau aggregation are briefly summarized. 1. Activation of tau-phosphorylating protein kinases. The glycogen synthase kinase GSK3ß phosphorylates tau and promotes its aggregation [32]. The kinase activity of GSK3ß is inhibited upon its phosphorylation by the cyclin-dependent protein kinase CDK5, which can also phosphorylate tau, thus either preventing or promoting tau aggregation, respectively [33]. The trimeric AMP-activated kinase complex AMPK phosphorylates and thus inhibits GSK3ß, but can also itself phosphorylate tau and promote its aggregation [34, 35]. The catalytic subunit of AMPK itself is activated by phosphorylation, which can be catalyzed either by the calmodulin-dependent protein kinase kinase CAMKK or a redundant pair of mitogen activated protein kinases (MAPKs), designated as extracellular signal recognition kinases ERK1 or ERK2 [36, 37]. Whilst CAMKK is activated by Ca2+-bound calmodulin, activation of ERK1/2 is triggered through a conserved MAPK cascade, consisting of the MAPKKs MEK1 and MEK2 and the MAPKKK RAF. Both branches are dependent on the function of the glutamate receptor NMDAR, which triggers an intracellular increase in calcium concentration. This results in the activation of CAMKK and indirectly of RAF through activation of a Ras-GDP/GTP exchange factor (GEF) not depicted here [38]. AMPK, and thereby its inhibitory effect on GSK3ß and tau aggregation, can also be activited by caloric restriction and antioxidants like resveratrol through their influence on the intracellular energy state, i.e. by changing the AMP/ATP ratio [39]. 2. Protein phosphatases. Only two major protein phosphatases which influence tau aggregation are depicted in this scheme, PP2A and calcineurin. Their roles in AD have been reviewed in [40]. Briefly, calcineurin, also called PP2B, activates the tau-kinase GSK3ß and promotes tau aggregation. It can also directly dephosphorylate specific residues in tau and has several other cellular targets, including the NMDAR receptor, relations that are not included here. PP2A has a large variety of substrates, only a few of which are depicted here. Interestingly, it has been found associated to microtubules and has been implicated in many aspects of AD, also reviewed in [40]. In this context, it can desphosphorylate protein kinases in signaling pathways leading to tau aggregation, such as ERK1/2 and AMPK, but also acts directly on phosphorylated tau. 3. Influence of Aß and mitochondrial functions on tau aggregation. The soluble forms of Aß are currently believed to trigger tau aggregation through signaling pathways which still need to be elucidated. Two possible connections are depicted in this scheme. Thus, Aß accumulation leads to an increased calcium flux through NMDAR and thus triggers both the CAMKK and the ERK1/2 mediated activation of AMPK [41]. While Ca2+ concentration and Aß accumulation are interdependent, the latter is prevented by the action of AMPK [42] and by the deacetylase SIRT1 [43]. SIRT1 also inhibits the formation of tau aggregates [44]. It should be noted that SIRT1 and AMPK activities are interdependent in that one can activate the other [100]. AMPK also activates the transcription factor PGC1α, which triggers mitochondrial biogenesis [45]. A by-product of mitochondrial respiration are reactive oxygen species (ROS), whose concentration increase with age and mitochondrial malfunctions. While this leads to aggregation of both Aß and tau, in turn these two pathological hallmarks of AD have been proposed to cause mitochondrial malfunctions [46, 47]. 4. Other protein kinases. Indirectly, several other protein kinas-es involved in different signaling pathways may influence tau aggregation. Thus, ULK1, which is activated by AMPK, promotes autophagy and thereby the degradation of tau aggregates. In the same physiological process, inhibition of the mTOR kinase complex, also mediated by AMPK, prevents its inhibitory effect on autophagy again promoting disposal of tau aggregates [48]. Finally, the death-activated protein kinase DAPK activates the protein isomerase PIN1, which acts on phosphorylated tau and restores its ability to interact with microtubules [49].
32. Hooper C, Killick R, Lovestone S (2008). The GSK3 hypothesis of Alzheimer’s disease. J Neurochem 104(6): 1433-1439. http://dx.doi.org/10.1111/j.1471-4159.2007.05194.x
33. Keeney JT, Swomley AM, Harris JL, Fiorini A, Mitov MI, Perluigi M, Sultana R, Butterfield DA (2012). Cell cycle proteins in brain in mild cognitive impairment: insights into progression to Alzheimer disease. Neurotox Res 22(3): 220-230. http://dx.doi.org/10.1007/s12640-011-9287-2
34. Park H, Kam TI, Kim Y, Choi H, Gwon Y, Kim C, Koh JY, Jung YK (2012). Neuropathogenic role of adenylate kinase-1 in Abeta-mediated tau phosphorylation via AMPK and GSK3beta. Hum Mol Genet 21(12): 2725-2737. http://dx.doi.org/10.1093/hmg/dds100
35. Vingtdeux V, Davies P, Dickson DW, Marambaud P (2011). AMPK is abnormally activated in tangle- and pre-tangle-bearing neurons in Alzheimer’s disease and other tauopathies. Acta Neuropathol 121(3): 337-349. http://dx.doi.org/10.1007/s00401-010-0759-x
36. Feld M, Krawczyk MC, Sol Fustinana M, Blake MG, Baratti CM, Romano A, Boccia MM (2014). Decrease of ERK/MAPK overactivation in prefrontal cortex reverses early memory deficit in a mouse model of Alzheimer’s disease. J Alzheimers Dis 40(1): 69-82. http://content.iospress.com/articles/journal-of-alzheimers-disease/jad131076
37. Spitzer P, Schieb H, Kamrowski-Kruck H, Otto M, Chiasserini D, Parnetti L, Herukka SK, Schuchhardt J, Wiltfang J, Klafki HW (2011). Evidence for Elevated Cerebrospinal Fluid ERK1/2 Levels in Alzheimer Dementia. Int J Alzheimers Dis 2011: 739847. http://dx.doi.org/10.4061/2011/739847
38. Thomas GM, Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci 5(3): 173-183. http://dx.doi.org/10.1038/nrn1346
39. Porquet D, Grinan-Ferre C, Ferrer I, Camins A, Sanfeliu C, Del Valle J, Pallas M (2014). Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer’s disease. J Alzheimers Dis 42(4): 1209-1220. http://content.iospress.com/articles/journal-of-alzheimers-disease/jad140444
40. Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC (2012). Protein phosphatases and Alzheimer’s disease. Prog Mol Biol Transl Sci 106:343-379. http://dx.doi.org/10.1016/b978-0-12-396456-4.00012-2
41. Danysz W, Parsons CG (2012). Alzheimer’s disease, beta-amyloid, glutamate, NMDA receptors and memantine–searching for the connections. Br J Pharmacol 167(2): 324-352. http://dx.doi.org/10.1111/j.1476-5381.2012.02057.x
42. Cai Z, Yan LJ, Li K, Quazi SH, Zhao B (2012). Roles of AMP-activated protein kinase in Alzheimer’s disease. Neuromolecular Med 14(1): 1-14. http://dx.doi.org/10.1007/s12017-012-8173-2
43. Godoy JA, Rios JA, Zolezzi JM, Braidy N, Inestrosa NC (2014). Signaling pathway cross talk in Alzheimer’s disease. Cell Commun Signal 12:23. http://dx.doi.org/10.1186/1478-811x-12-23
44. Julien C, Tremblay C, Emond V, Lebbadi M, Salem N, Jr., Bennett DA, Calon F (2009). Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol 68(1): 48-58.http://dx.doi.org/10.1097/nen.0b013e3181922348
45. Pedros I, Petrov D, Allgaier M, Sureda F, Barroso E, Beas-Zarate C, Auladell C, Pallas M, Vazquez-Carrera M, Casadesus G, Folch J, Camins A (2014). Early alterations in energy metabolism in the hippocampus of APPswe/PS1dE9 mouse model of Alzheimer’s disease. Biochim Biophys Acta 1842(9): 1556-1566. http://dx.doi.org/10.1016/j.bbadis.2014.05.025
46. Eckert A, Nisbet R, Grimm A, Gotz J (2014). March separate, strike together–role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer’s disease. Biochim Biophys Acta 1842(8): 1258-1266.http://dx.doi.org/10.1016/j.bbadis.2013.08.013
47. Luque-Contreras D, Carvajal K, Toral-Rios D, Franco-Bocanegra D, Campos-Pena V (2014). Oxidative stress and metabolic syndrome: cause or consequence of Alzheimer’s disease? Oxid Med Cell Longev 2014:497802. http://dx.doi.org/10.1155/2014/497802
48. Alers S, Loffler AS, Wesselborg S, Stork B (2012). Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 32(1): 2-11. http://dx.doi.org/10.1128/mcb.06159-11
49. Kim BM, You MH, Chen CH, Lee S, Hong Y, Kimchi A, Zhou XZ, Lee TH (2014). Death-associated protein kinase 1 has a critical role in aberrant tau protein regulation and function. Cell Death Dis 5:e1237. http://dx.doi.org/10.1038/cddis.2014.216