
Reviews:
Microbial Cell, Vol. 6, No. 3, pp. 142 - 159; doi: 10.15698/mic2019.03.670
Microevolution of the pathogenic yeasts Candida albicans and Candida glabrata during antifungal therapy and host infection

1 Department of Bioengineering, Instituto Superior Técnico, Universidade de Lisboa, Lisboa, Portugal.
2 iBB – Institute for Bioengineering and Biosciences, Biological Sciences Research Group, Instituto Superior Técnico, Lisboa, Portugal.
# Contributed equally.
Keywords: fungal pathogens, host-pathogen interaction, microevolution, virulence, biofilm formation, antifungal resistance.
Received originally: 15/01/2019 Received in revised form: 30/01/2019
Accepted: 01/02/2019
Published: 08/02/2019
Correspondence:
Dr. Miguel C Teixeira, IBB - Institute for Bioengineering and Biosciences, Biological Sciences Research Group, Instituto Superior Técnico, University of Lis-bon, Av. Rovisco Pais, 1049-001 Lisboa, Portugal; Tel. +351-218417772; Fax +351-218419199; mnpct@tecnico.ulisboa.pt
Conflict of interest statement: The authors declare no competing interests.
Please cite this article as: Pedro Pais, Mónica Galocha, Romeu Viana, Mafalda Cavalheiro, Diana Pereira, Miguel Cacho Teixeira (2019). Microevo-lution of the pathogenic yeasts Candida albicans and Candida glabrata during antifungal therapy and host infection. Microbial Cell 6(3): 142-159. doi: 10.15698/mic2019.03.670
Abstract
Infections by the pathogenic yeasts Candida albicans and Candida glabrata are among the most common fungal diseases. The success of these species as human pathogens is contingent on their ability to resist antifungal therapy and thrive within the human host. C. glabrata is especially resilient to azole antifungal treatment, while C. albicans is best known for its wide array of virulence features. The core mechanisms that underlie antifungal resistance and virulence in these pathogens has been continuously addressed, but the investigation on how such mechanisms evolve according to each environment is scarcer. This review aims to explore current knowledge on microevolution experiments to several treatment and host-associated conditions in C. albicans and C. glabrata. The analysis of adaptation strategies that evolve over time will allow to better understand the mechanisms by which Candida species are able to achieve stable phenotypes in real-life scenarios, which are the ones that should constitute the most interesting drug targets.
INTRODUCTION
Infections by fungal pathogens have become a relevant health problem, especially for the increasing immunocompromised population [1]. Infections by Candida species are the most common cause of fungal infections and represent the 4th leading cause of hospital acquired bloodstream infections in the USA [2][3][4]. Candida albicans and Candida glabrata represent the two most commonly isolated species worldwide [2][5].
–
Despite representing the bulk of Candida infections, each species possesses quite different traits in terms of antifungal susceptibility profiles and virulence features. C. glabrata presents high levels of intrinsic and acquired resistance to azole antifungals, especially due to overexpression of multidrug resistance transporters activated by the transcription factor Pdr1 [6][7][8][9]; while C. albicans isolates are usually more susceptible to azole treatment [10]. On the other hand, C. albicans carries a number of virulence features that are absent in C. glabrata, such as the formation of hyphae. Hyphal formation plays an important part in colonization and biofilm formation, which is consistent with the notion that C. albicans biofilms are bulkier than the ones formed by C. glabrata [11]. Moreover, hyphae contribute for tissue invasion and phagocyte escape [12][13][14][15]. C. glabrata mechanisms of tissue invasion are mostly unknown; although it is hypothesized to possibly occur by endocytosis induction of host cells [16]. As for phagocyte escape, C. glabrata applies a persistence strategy by replicating inside phagocytes and eventually leading to cell lysis due to fungal load [17][18], rather than actively escaping. The production of secreted aspartyl proteases (SAPs) is another critical virulence trait in C. albicans, allowing the pathogen to degrade host barriers and invade surrounding tissue [19]. In turn, C. glabrata does not appear to produce significant levels of proteinase activity [20] nor to induce significant tissue damage [16]. However, C. glabrata possesses a family of aspartic proteases, which is mainly associated with cell wall remodeling and possible immune evasion [21]. In addition, the expression of phospholipases is yet another feature that allows C. albicans to acquired nutrients in host nutrient-poor niches and contributes to invasion, whereas C. glabrata shows a very low level of phospholipase activity [20].
–
This review aims to explore the data retrieved from microevolution experiments performed on both C. albicans and C. glabrata. Comprehensive knowledge about the mechanisms of antifungal resistance and virulence, their clinical prevalence and strategies to counteract them has been duly explored. However, the way such mechanisms evolve during colonization of the human host and upon confrontation with antifungal treatment can provide valuable insight on the pathogenic nature of Candida spp. employed in the clinical setting. By better understanding the way Candida spp. evolve in distinct environments and selective pressures, it could be possible to delineate better strategies to tackle infections by these pathogens.
CANDIDA EVOLUTION TOWARDS DRUG RESISTANCE
Antifungal drugs and resistance mechanisms in C. albicans and C. glabrata
–
Azoles are the common first-line drugs against most Candida species because of their safety profile and availability in both oral and intravenous formulations [22]. They act by inhibiting the 14α-demethylase Erg11 in the ergosterol biosynthesis pathway and cause the accumulation of the toxic sterol 14,24-dimethylcholesta-8,24(28)-dien-3β,6α-diol (DMCDD) that permeabilizes the plasma membrane [23]. Nevertheless, the fungistatic nature of azoles imposes strong directional selection for the evolution of resistance. Additionally, some Candida species, such as C. glabrata, are intrinsically less susceptible to this class of antifungal drugs. In fact, C. glabrata has risen dramatically in frequency as a significant cause of blood stream infection (BSI) since the introduction of azole drugs in the 1980s [24]. The increase in the prophylactic use of azoles for high-risk individuals undoubtedly contributed to the increasing development of C. glabrata resistance to these antifungal drugs, which are significantly effective in eradicating infections caused by other Candida species [25][26][27]. Still, these anti-fungals are inactive against biofilm-associated infections, which is a significant public health problem due to the increasing usage of medical devices [28].
–
C. albicans might develop resistance toward azoles through upregulation of efflux pumps Cdr1, Cdr2 and Mdr1, inactivation of Erg3 that synthesizes the toxic sterol DMCDD, and upregulation or mutations in the gene encoding azoles target, ERG11 [29][30]. Generally, the upregulation of drug efflux pumps and drug target is the result from point mutations in genes encoding the regulators of their expression [31][32][33][34][35][36], or from increased copy number of the genes through genome rearrangements such as whole chromosome and segmental aneuploidies [37][38][39]. Moreover, it was very recently demonstrated that C. albicans can also gain azole resistance by altering sphingolipid composition, in vitro [40].
–
In contrast to what is observed in C. albicans and despite the potential for ERG11 point mutations to have a greater impact in haploid organisms, as is the case of C. glabrata, several studies suggest that mutations in ERG11 are not involved in clinical azole resistance in this pathogen [8][41][42]. The major described mechanism of acquired azole resistance in C. glabrata clinical isolates is the increased drug efflux due to the upregulation of drug efflux pumps [43][44][45][46]. This is generally caused by gain-of-function (GOF) mutations within the gene encoding the key transcriptional regulator of drug resistance, PDR1, responsible for the upregulation of the drug efflux pumps, Cdr1, Cdr2, Snq2 and Qdr2, which directly confer most of the acquired azole resistance in this pathogen and, surprisingly, was also found to enhance virulence [41][46][47][48]. A high frequency of acquired azole resistance in vitro in C. glabrata populations has been linked to a loss of mitochondrial function, which leads to the upregulation of ABC transporter genes [47][49]. In fact, this phenotype is associated with Pdr1 expression, as mitochondrial dysfunction was shown to increase the expression of PDR1, further exacerbated by a positive auto-regulatory loop that leads to PDR1 and target genes overexpression [6][50]. It was proposed that this pathogen can switch between states of mitochondrial competence (azole-susceptible) and incompetence (azole-resistant) in response to azole exposure, probably through chromatin epigenetic modifications [51]. Until recently, clinical relevance of mitochondrial mutants was questionable in light of their decreased fitness. Nevertheless, Ferrari et al. found that an azole-resistant C. glabrata clinical isolate not only exhibited mitochondrial dysfunction and upregulation of CDR1 and CDR2, but also was more virulent than its susceptible counterpart in both systemic and vaginal murine infection models [52]. This report demonstrated that mitochondrial dysfunction can confer selective advantage under host conditions. Calcium signaling was also proposed to play a role in azole resistance, since the loss of Ca2+ signaling pathway changes fluconazole activity from fungistatic to fungicidal in vitro [51]. Furthermore, in very recently published data, at least 78 other genes were suggested to be involved in C. glabrata resistance toward fluconazole and voriconazole [53]. This points to the hypothesis that there might be other yet unknown alternative paths to azole acquired resistance in this pathogen.
–
Echinocandins are the only new class of antifungals to reach the clinic in decades, with three echinocandins currently available for clinical use: caspofungin, micafungin, and anidulafungin [26][30]. These antifungals target the fungal cell-wall by acting as noncompetitive inhibitors of β-(1,3)-D-glucan synthase enzyme complex which catalyzes the production of glucan, the major component in Candida cell walls [26][54]. The disruption of (1,3)-β-D-glucans impairs the structure of growing cell walls, resulting in loss of structural integrity, osmotic instability and cell death. Therefore, a fungicidal effect is accomplished by disrupting cell wall synthesis. More important, not only echinocandins present good safety profiles and their toxicity is very low due to their unique target, that is absent in mammalian cells, but also have been shown to have activity against Candida biofilms as the inhibition of polysaccharide production could lead to lysis and dissolution of the extracellular matrix (ECM) [55][56]. This is an extremely important feature since biofilm-associated infections are very hard to treat and are recurrent in patients with medical devices such as pacemakers or catheters. Considering all the advantages, the Infectious Diseases Society of America guidelines currently favor echinocandins as first-line treatment for systemic candidiasis in patients with moderate-to-severe infection and in those with prior exposure to azoles, with fluconazole held in reserve for the treatment of patients with less severe infections [57]. Moreover, the European Society of Clinical Microbiology and Infectious Diseases recommends echinocandins as first-line treatment for all patients with systemic candidiasis [58]. Even so, Candida clinical isolates exhibiting reduced echinocandin susceptibility have been found over the past years [59][60][61]. In C. albicans, this reduced susceptibility is strictly attributed to changes in the subunit of glucan synthase enzyme complex Fks1, which is the specific target of echinocandins. Indeed, genetically related isolates from the same patient with different mutations in FKS1 were identified, suggesting that reduced susceptibility can evolve in the patient [60][62]. Otherwise, mutations in both Fks1 and its paralog Fks2 (but not Fks3) have been associated with resistance in C. glabrata [63]. Interestingly, recent studies reported that echinocandin resistance is more common in C. glabrata compared with other species and that this rate can be attributed to the high potential of C. glabrata for developing resistance mutations [64][65]. In 2011, Costa-de-Oliveira et al. reported for the first time the in vivo acquisition of echinocandin resistance following anidulafungin therapy in a patient with C. glabrata invasive candidiasis, highlighting the need for antifungal susceptibility surveillance in patients under extended echinocandin therapy [61].
–
Polyenes, such as amphotericin B (AmB), are fungicidal drugs that act by binding to ergosterol in the lipid layer. The typical mode of action associated with polyenes is the resulting formation of pores in the cell membrane, ultimately leading to fungal cell permeabilization [23]. Additionally, merely the binding of AmB to ergosterol was shown to kill yeast cells, with pore formation being a complementary mechanism [66]. These antifungals have the broadest spectrum of activity compared to any other anti-fungal molecules. However, polyenes are highly toxic since they have a lower but non-negligible affinity for cholesterol, the mammalian membrane sterol, which is responsible for numerous side effects. These side effects of therapy with polyenes, namely AmB, are substantial and may be divided into acute (fever, vomiting, headache) and sub-acute (kidney problems or failure) [67]. Despite the noteworthy disadvantages, AmB is still used for the treatment of the most serious fungal infections, in large part due to its broad spectrum of activity. However, resistance toward this anti-fungal has been found in many clinical isolates, including C. glabrata [68][69][70]. The molecular mechanisms underlying polyene resistance are poorly documented, especially in pathogenic yeasts. It is thought that a decrease in the levels of ergosterol in the cell membrane is connected to the resistant phenotype [70][71].
–
Pyrimidine analogs such as flucytosine were first used to treat fungal infections in the 1960s. These antifungals have fungistatic activity exerted through the interference with pyrimidine metabolism, as well as RNA/DNA and protein synthesis. Yet, the quick inception of resistance banned the use of flucytosine as a monotherapy, and consequently it is only used in combination with other anti-fungals [72]. Decreased susceptibility to the most used pyrimidine, flucytosine, has been related with point mutations in FUR1, FCY1 (encoding enzymes involved in the pyrimidine pathway) and FCY2 (encoding a cytosine permease) genes in both C. albicans and C. glabrata [73][74]. Moreover, it was recently discovered in our lab that the deletion of FPS1 or FPS2, encoding aquaglyceroporins, leads to an increased accumulation of flucytosine within C. glabrata cells [75].
–
Considering the intrinsic variation regarding drug susceptibility among different Candida species and the increasing acquired resistance in several clinical isolates, it becomes evident that the genetic evolution underlying drug resistance phenomenon must continue to be investigated. Unveiling new potential targets is imperative to develop new suitable strategies to fight increasing Candida-associated infections and, although the nature of intrinsic resistance is unknown, the development of resistance can be studied.
–
Evolution of C. albicans and C. glabrata resistance toward antifungal drugs
–
The acquisition of resistance depends on various factors and differs severely between species, cell populations and the imposed stress. Genome dynamics and evolution according to the cell’s needs is the process through which cells are able to adapt and survive in hostile environments, such as the presence of drugs. Evolutionary changes in organismal traits may occur either gradually or suddenly and encompasses single-point mutations; gene duplications, deletions, inversions, and insertions; chromosomal rearrangements; aneuploidies; the loss of heterozygosity (LOH, in diploid organisms); and horizontal gene transfer and/or hybridization [76]. Noteworthy, recent advances in DNA sequencing technologies have now made it possible to identify genetic changes between ancestral and derived organisms on a whole-genome scale for any species. Therefore, adaptative evolution experiments can provide insights into both the genetic basis and dynamics of adaptation to a specific environment.
–
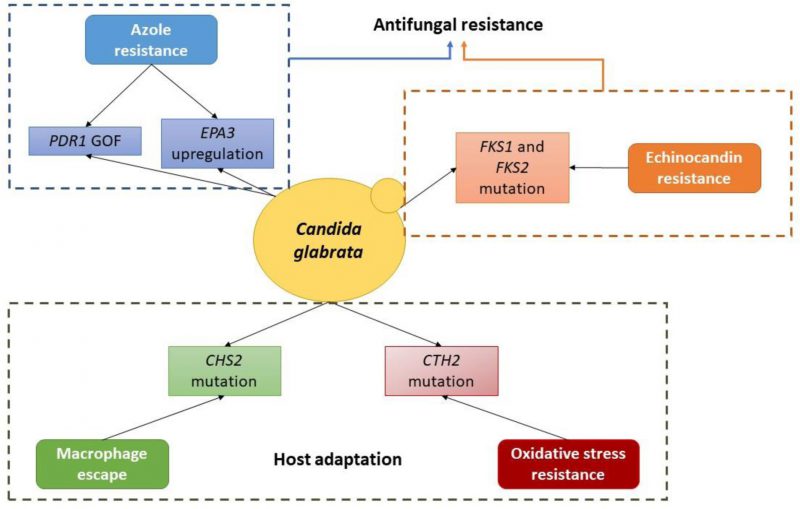 |
FIGURE 1 Schematic depiction of the currently known molecular basis of evolution towards antifungal resistance and host adaptation in C. glabrata. |
–
The emergence of adaptative mutations is unpredictable and poses genetic variability within the cell population. Genetic adaptation is especially rapid when microbial populations are introduced into new environments [77]. For instance, antifungal treatment exerts selective pressure to which only the cells that acquire resistance-related mutations will be able to survive. However, fungistatic drugs favor the occurrence of resistance-related mutations rather than fungicidal ones. The fact that these drugs do not lead to cell death, instead their action results in growth arrest, allied to the high genome plasticity of Candida species enables cells to adapt to the new hostile environment through genetic modifications. Afterwards, selective pressure will select those that acquired resistance mutations to survive and give rise to resistant progeny therefore propagating the resistant phenotype in the population. Likewise, the drug-dosage regime is an important feature that influences the rate of resistance achievement. It was seen in vivo that less frequent administration of high drug dosages leads to faster emergence of resistance rather than the application of more periodic lower dosages [78]. Nonetheless, regardless of the rate at which resistance to the different antifungal classes arises, resistance to all classes of antifungals has been reported in both the laboratory and clinical settings [26]. Occasionally, within a clonal cell population, there is significant variability in the response to anti-fungals. This is called heteroresistance and constitutes a very important trait of pathogenic fungal populations as heteroresistant strains are not detected in standard susceptibility assays and may be a driver of azole resistance and therapy failure [79].
–
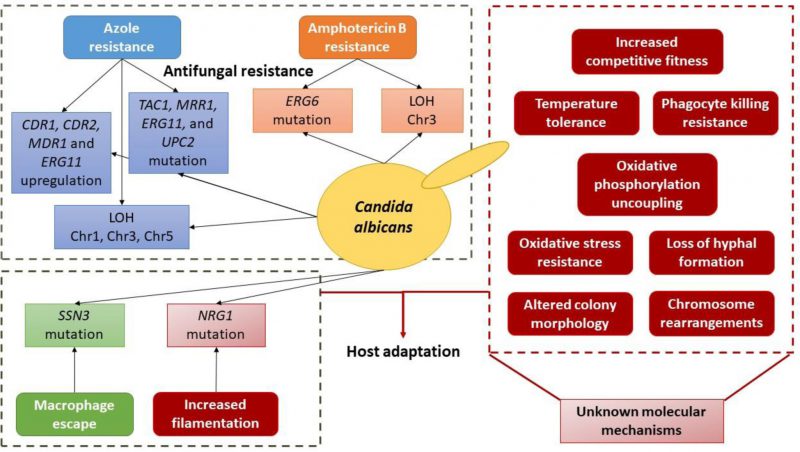 |
FIGURE 2 Schematic depiction of the currently known molecular basis of evolution towards antifungal resistance and host adaptation in C. albicans. |
–
In vivo studies of sequential clinical isolates to monitor the evolution of antifungal resistance complemented by in vitro experimental evolution is a powerful approach to unveil the mechanisms underlaying the development of antifungal resistance in different species against specific antifungals in a timescale. This allied with next-generation sequencing (NGS) and comparative genomics not only enable the detection of footprints of genetic evolution, but also are powerful tools to unveil the genomic changes accompanying the emergence of resistance. Some studies probing azole and echinocandin resistance mechanisms in the clinical setting have shown the existence of resistant isolates that do not display typical resistance hallmarks (e.g., PDR1 GOF or FKS mutations) [80][81][82]. Microevolution studies can contribute to better elucidate the evolution of additional resistance mechanisms that can occur in the clinic environment. Furthermore, these studies can provide a way to investigate population dynamics and hetero-resistance, what would add value to current knowledge on how initial and transient resistance mechanisms arise.
–
Evidences of resistance evolution: in vitro
–
In vitro evolution experiments provide a framework for understanding genome-wide changes that occurred during cell adaptation. Although the clinical relevance of in vitro studies is questionable in light of the feeble mimicking of human host environment, they have several associated advantages: controlled experimental conditions make the processes reproducible and facilitate the identification of factors that drive drug resistance; it is possible to have real-time control of new emergent features; easy replication allows the sample size to be greater and so genetic drift is minimized, since the population size at each transfer must be large enough to avoid beneficial mutations that arise to be lost to random drift [77]. These in vitro evolution experiments coupled with whole genome sequence techniques constitutes a powerful tool to address acquired resistance phenomena as it is possible to have a genome-wide view of adaptation and drug resistance progress and establishment.
–
In vitro evolution experiments might be carried out following a serial dilution system or a continuous system. The serial dilution system is the most used method due not only to its lower cost, but also to the feasibility in conducting an experiment that requires such a high number of replicates in parallel. Routinely, cells are transferred by serial dilution of a stationary-phase culture into fresh medium containing a concentration of drug that is inhibitory but not lethal [83]. For instance, Cowen et al. monitored the development of resistance toward fluconazole in replicated experimental populations founded from a single, drug-sensitive C. albicans cell for 330 generations [83]. Their results demonstrated that not only all populations exposed to fluconazole were able to adapt to its presence, but also that the development of drug resistance followed different trajectories in the initially identical populations. The expression profile of genes known to be involved in azole resistance (CDR1, CDR2, MDR1 and ERG11) was different within the resistant populations, as well as the level of resistance. Moreover, other genomic changes such as alterations in DNA fingerprints were detected in the resistant populations, although their connection with drug resistance is not known. Overall, the authors concluded that chance, in the form of mutations that confer an adaptive advantage, is a determinant in the evolution of drug resistance in experimental populations of C. albicans.
–
C. albicans resistance development toward the fungicidal AmB has also been studied in vitro. Vincent et al. sequenced sequentially laboratory evolved strains and observed that mutations in ERG6 followed by loss of heterozygosity of chromosome 3, where ERG6 resides, were behind AmB resistance acquisition [84]. Nevertheless, this acquired resistance came at a great cost. AmB-resistant mutants were hypersensitive to oxidative stress, febrile temperatures, and killing by neutrophils and had defects in filamentation and tissue invasion. Furthermore, the authors saw that these strains were avirulent in a mouse infection model. Despite the fact that costs of evolving resistance to AmB limit the emergence of this phenotype in the clinic, resistant clinical isolates of both C. albicans and C. glabrata have been found over the past years [68][69][70].
–
Another example of in vitro evolution experiment following a serial dilution system is a very recent study performed in our lab in which an in vitro evolution of a C. glabrata isolate toward multiazole resistance was carried out. Cavalheiro et al. analyzed the changes in the genetic profile of a C. glabrata isolate from azole susceptibility to posaconazole, clotrimazole, fluconazole and voriconazole resistance, evolved in a stepwise manner induced by longstanding incubation with fluconazole [85]. Remarkably, despite all the evolved populations presented reduced azole accumulation when compared to the parental one, only that displaying resistance toward all azoles acquired a GOF mutation in PDR1 and therefore upregulation of multidrug resistance transporter genes. Even more interesting was the correlation of drug resistance with increased expression of adhesin genes, namely EPA3. The authors demonstrated that the intermediate population displaying resistance toward posaconazole and clotrimazole had increased expression of adhesins and, consequently, an increased ability to adhere to other C. glabrata cells and to epithelial cells. Specifically, they showed that the absence of Epa3 adhesin increases C. glabrata cells susceptibility to all azoles tested. Epa3 and possibly other adhesins, were proposed to play a role in azole resistance probably by promoting cellular aggregation, which protects the cells from extracellular drug concentration. Interestingly, the overexpression of adhesin-like genes in C. glabrata resistant isolates was demonstrated in vivo by others [86][87], but their role in drug resistance was never analyzed until this study that enhances the presence of yet unknown mechanisms driving acquisition of resistance. It was suggested that azole-exposed C. glabrata population is iteratively selected toward resistance at minimum cost. Full resistance seems to be acquired through the emergence of PDR1 GOF mutations and, consequently, overexpression of drug efflux pumps. However, before this, the population transcriptome is remodeled reflecting the transient selection of more-fit subpopulations. When PDR1 GOF mutations emerge in part of the population, natural selection acts favoring these more-fit cells and ultimately leading to the disappearance of other subpopulations.
–
Very recently, Bordallo-Cardona et al. investigated the ability of five echinocandin-susceptible C. glabrata isolates to acquire in vitro resistance to anidulafungin and micafungin and, remarkably, all isolates acquired resistance after 2–4 days of exposure to low and constant micafungin concentrations and mutations in FKS2 were found in all of them [88]. This study highlights the idea that the constant exposure to low doses of echinocandins promotes the development of resistance. Moreover, these results showed the ease of resistance acquirement, even to fungicidal drugs, stressing the need for the development of new anti-fungals to eradicate Candida infections, or the design of drugs that target resistance mechanisms.
–
In contrast with the most used serial dilution system, the continuous culture system implies that the environment and the cell physiological state are maintained constant throughout the course of the experiment. Huang et al. reported the first study to investigate the emergence of adaptative traits in C. albicans during in vitro evolution in continuous cultures under increasing concentrations of fluconazole [89]. They used a visualizing evolution in realtime (VERT) approach that not only enables the monitoring of adaptative events, but also facilitates the identification and isolation of adaptative mutants from the population [90]. Some remarkable findings were that several resistant mutant cells appeared rapidly in independent lineages and that a higher frequency of adaptative events occurred in the presence of the drug. Moreover, data shown in this study sustain the idea that rapid resistance can arise from mutations in subpopulations and also suggest that drug resistance mechanisms are not always related with fitness cost.
–
Evidences of resistance evolution: in vivo
–
The most reliable approach to study the evolutionary emergence of drug resistance involves the analysis of isolates from an infected individual over treatment time. However, although this approach is the most clinically relevant, it has some significant constrains such as the small sample sizes. Due to the limited number of clones isolated, the population dynamics during the emergence of drug resistance in vivo has been difficult to assess [89]. Also, population parameters such as the genotype of the initial population or the exact number of generations are hard to determine.
–
Despite these constrains, many efforts have been made to understand the emergence of resistance in fungal pathogens in vivo, namely in Candida species. More than two decades ago, White et al. analyzed a set of 17 isolates of C. albicans collected over two years from an HIV-infected patient who was receiving azole treatment for recurrent oropharyngeal candidiasis [91]. The initial isolate in the series was susceptible to fluconazole treatment, but by the end of the two-year period, the strain had acquired a level of resistance 200-fold higher than that of the original isolate. Over the time of sampling, mutations were acquired in the gene encoding the fluconazole target, ERG11 [92][93]. Noteworthy, the overexpression of the CDR1 and MDR1 efflux pumps was also detected, although the causes were not evaluated in this study [91]. Ten years later, another study addressing this series of isolates (among others) identified GOF mutations in the transcription factor TAC1 [94], one of which was identified more recently in another isolate as well [95]. The same study also identified GOF mutations in MRR1 and LOH of the mutated allele was associated with the observed Mdr1 overexpression in these isolates [95]. Yet another study, by Cowen et al., used this same set of C. albicans isolates to evaluate the impact of the essential chaperone Hsp90 in the natural evolutionary process [96]. Interestingly, they found that inhibition of Hsp90 or of calcineurin decreased fluconazole resistance, more in early isolates rather than in later ones. Drugs structurally related to GdA (Hsp90 inhibitor) are currently undergoing clinical evaluation [97] and present a good alternative to prevent de novo evolution of resistance. Hsp90 inhibitors are effective in overcoming fungal drug resistance at concentrations that are clinically well tolerated. In 2011, Hoot et al. analyzed the sequence of the transcriptional regulator UPC2, that regulates the expression of ERG11, in the same 17 isolates from the set and searched for mutations [31]. They found a mutation in UPC2 that caused the overexpression of ERG11 contributing for the resistant phenotype [31]. Previous studies probing azole resistance in matched fluconazole-resistant and susceptible C. albicans isolates had also described the occurrence of UPC2 GOF mutations that result in the overexpression of ERG11 and consequent azole resistance [31][32].
–
In 2006, Coste et al. matched pairs of azole-susceptible and azole-resistant C. albicans clinical isolates were screened for mechanisms that could explain azole resistance [35]. The authors found that GOF mutations in the azole resistance regulator TAC1, followed by LOH, was responsible for azole resistance [35]. In 2007, Coste et al. analyzed the genetic and genomic events that lead to azole resistance through alteration of TAC1 and ERG11 in sequential clinical isolates [94]. Consistent with previous reports addressing the acquisition of azole resistance [91][98], the development of azole resistance in C. albicans was proposed to occur in a stepwise manner, in which the acquisition of mutations in drug resistance genes, such as TAC1 and ERG11, is a prerequisite for the development of azole resistance and is followed by different LOH events, including chromosome loss [94]. These chromosome alterations can be accompanied by acquisition of extra chromosomal elements (isochromosome formation), all contributing to drug resistance in C. albicans. The authors proposed that not only the presence of specific mutations in azole resistance genes is necessary but also their increase in copy number by LOH and the addition of extra Chr5 identical copies. Likewise, it was reported that mutations in the multidrug resistance regulator MRR1 and LOH are the main cause of the multidrug transporter Mdr1 overexpression in fluconazole resistant C. albicans strains [34][99].
–
In 2015, Ford et al. studied the genetic base of yeast adaptation to drug treatment in the human host using whole-genome sequencing of sequential isolates from the same patient [100]. 43 C. albicans isolates were taken over time from eleven oral candidiasis patients who were being treated with fluconazole and their genome was sequenced demonstrating frequent LOH and single nucleotide polymorphisms (SNPs) in 166 genes as the main modifications associated with decreased fluconazole susceptibilities. Both chromosome 3, which comprises CDR1 and CDR2 (efflux pumps coding genes) and MRR1 (MDR1 efflux pump regulator), and chromosome 5, which contains ERG11 and TAC1 (positive regulator of CDR1 and CDR2), were found to suffer LOH. Even more interesting, the authors found that mutations in cell adhesion, filamentous growth and biofilm formation related genes occurred in several isolates, which suggests a co-evolution between virulence and resistance. Moreover, a lot of genetic variation was found between samples from the same patient, demonstrating the selective pressure acting on the population.
–
Recently, Vale-Silva et al. analyzed the genome of two C. glabrata clinical isolates taken from the same patient, one before (azole susceptible) and another after (azoleresistant) a time-lapse of 50 days of azole therapy [86]. Only 17 nonsynonymous SNPs were found comparing the genomes of both strains, among which a known PDR1 GOF mutation, and small size indels in coding regions mostly in adhesin-like genes. Interestingly, both isolates carried a nonsynonymous mutation in MSH2 known to favour the hyper-mutator phenotype (V239L) and the number of accumulated mutations between both clinical isolates was shown to be consistent with the presence of a MSH2 defect [101]. Nonetheless, the remaining genetic alterations were attributed to fitness or accidental mutations and not to the resistant phenotype, which was exclusively attributed to the PDR1 GOF mutation. Interestingly, C. glabrata virulence and adherence to host cells was also linked to the emergence of PDR1 GOF mutations in clinical isolates. PDR1 GOF mutations have been associated with increased C. glabrata virulence [48], increased expression of adhesins and increased adherence to host epithelial cells [87][102].
–
In 2012, Singh-Babak et al. reported the first genomewide analysis of mutations occurring during evolution of echinocandin resistance in a series of C. glabrata isolates recovered over a 10-month period from a patient that underwent caspofungin treatment for bloodstream candidemia [103]. Although a nonsynonymous mutation in the drug target gene FKS2 is enough for the resistant phenotype, an elevated fitness cost was associated with it. Remarkably, further acquired mutations demonstrated to be able to mitigate the fitness cost caused by the FKS2 mutation. The authors also implicate the molecular chaperone Hsp90 and calcineurin in the resistant phenotype acquired by the FKS2 mutation [103], however, the inhibition of Hsp90 in C. glabrata, as well as in C. albicans, decreases basal tolerance to drugs [96][103][104][105], and so, a particular role of this pathway in the evolution and acquisition of antifungal resistance should be address with caution.
–
Otherwise, Biswas et al. performed a genome-wide analysis of C. glabrata antifungal resistance markers to simultaneously unveil mutation patterns of genes known to be involved in resistance toward different drug classes [106]. NGS was used to unravel SNPs between resistant strains and susceptible counterparts. Although high-quality nonsynonymous SNPs were found in FKS1 and FKS2 (echinocandin resistance), FCY2 (flucytosine resistance), PDR1 (azoles resistance), the later were found in both azolesusceptible and resistant strains, highlighting the need for further investigation.
CANDIDA EVOLUTION TOWARDS INCREASED BIOFILM FORMATION
Biofilms are organized communities of cells developed after adhesion to a surface and enclosed in an ECM [107][108]. The ability to grow as biofilms presents an advantage for fungal pathogens, regarding colonization and persistence in different host tissues [109], but also in the resilience against antifungal treatments [110][111]. This is especially true for C. albicans and C. glabrata. Both species are known to use biofilms to colonize the surface of several medical devices based on different materials [112]. As a result, biofilm formation comes with strong impact in medicine as well as in the development of improved therapeutic solutions.
–
Biofilm formation is a multifaceted process well described for C. albicans in several in vitro studies [109][112]. It develops through four distinct steps: the early phase, characterized by the adhesion of yeast cells to a surface; the intermediate phase, consisting in cell proliferation and differentiation into pseudohyphae and hyphae, accompanied by the production and secretion of extracellular polymeric substances (EPS); the maturation phase, in which fully-formed three-dimensional biofilm structure surrounded by a thick ECM is reached; and the dispersal phase, where detachment of round budding yeast cells from the biofilm may lead to the formation of biofilms in other host niches [112][113][114]. Although very simply described herein, the formation of Candida biofilms is a very complex process which relies on different mechanisms, such as cell-to-cell adhesion, crucial for the integrity of the biofilm [113], or the formation of the ECM, composed by different EPS, some identified as important for the resistance to antifungal drugs [115].
–
To tackle C. albicans and C. glabrata biofilms from a therapeutic point of view, it is necessary to identify which of these mechanisms, responsible for the formation and maintenance of the biofilm, are used in vivo to persist in the human host or on medical devices, and if they change overtime during the infection of a given patient, increasing the strain’s ability to form bigger, thicker or more stable biofilms. Although few information has been collected regarding these topics, some studies have been focused on biofilm forming ability of Candida sequential isolates.
–
Jain et al. assessed the biofilm forming ability of ten sequential urine isolates available from nine patients, taken in a time-range of 70 days [116]. The isolates collected from four of the nine patients were composed of both C. albicans and C. glabrata species. The single strain isolates revealed no significant variability in biofilm forming ability, even comparing isolates taken with 70 days in between. However, it varied in infections caused by more than one strain [116]. In line with these results, another study reported biofilm forming ability as a specific trait of a given Candida isolate that remains stable in serial isolates [117]. Moreover, Bitar et al., from 85 clinical isolates obtained from different patients, have identified by multilocus sequence typing (MLST) two isolates that shared homology features, suggesting a possible case of microevolution of one strain that may have spread into another patient. Once again, no variability in the capacity to form biofilms was observed between the two isolates [118]. Although it seems that biofilm formation ability of an isolate does not change often throughout the infection time in a given patient, there are some cases of registered evolution [116], but there is no information on the underlying differences in genome sequence. More studies should be performed to assess these changes that may underly important features of host adaptation. It is important to note that although a given strain does not appear to change its ability to form biofilm over the course of an infection, biofilm formation among distinct isolates is quite disparate. The mechanisms underlying distinct biofilm formation ability across isolates is largely unknown and warrants further investigation.
–
In addition, it would be interesting to try to evolve the capacity of Candida strains to form more biofilm. In vitro evolution of Candida strains towards higher biofilm formation would help identify the main factors influencing biofilm formation, as well as the more propitious environment (materials and in vivo models) for Candida biofilms. The study of Candida strains in various time-points of biofilm formation and maintenance would enhance our knowledge about the more essential mechanisms for Candida biofilms and their persistence and evolution in the human host.
CANDIDA EVOLUTION TOWARDS ADAPTION TO HOST NICHES
Microevolution plays an important role on the remarkable capacity of adaptation of Candida species to the many different host niches. Evolution experiments have emerged as a powerful tool for testing specific theoretical models and are a great tool for understanding microevolution and to study factors which are important for local adaptation and for the survival of the pathogens in the host [119][120].
–
There are already some studies on microevolution of C. albicans in serial passage experiments through different host niches, namely kidney [121] and spleen [121][122], two main target organs in murine Candida sepsis [123]. In Lüttich et al., serial passage experiments of the commonly used C. albicans strain SC5314 through the murine kidney, showed that despite leading to increased phenotypic variability within the population possibly by microevolution, the overall virulence, fungal fitness and the host response did not follow a clear trend between infected animals, revealing that the C. albicans strain used was already well adapted to the murine kidney [124].
–
However, in another case, serial passage experiments with the same C. albicans strain through murine spleens were performed and at the fifth passage a stable respiratory mutant isolate was recovered, showing delayed filamentation initiation and abnormalities in carbon-assimilation. This recovered strain was more resistant to phagocytosis by neutrophils and macrophages and showed attenuated virulence in mice. It was hypothesized that this mutant seems to persist and proliferate without killing the mice. The regulation of respiration was proposed to have influence in the interaction between C. albicans and the host, as the mutant displayed uncoupled oxidative phosphorylation, a possible mechanism of adaptation to stressful host environments by C. albicans [121].
–
In Forche et al., genome wide genetic and phenotypic evolution were evaluated in C. albicans in two different scenarios: in vivo, during a passage through a murine host and in vitro, during propagation in liquid culture. Populations passaged in vivo showed slower growth and higher rates of chromosome-level genetic variation compared to those grown in vitro [122]. These results seem to indicate that a passage through a living host may lead to changes in the C. albicans competitive fitness and seems to result in slower growth and higher rates of genomic and phenotypic variation compared to in vitro populations. In fact, recently, Tso et al. demonstrated that C. albicans can be experimentally induced to increase its competitive fitness in the gut of a mouse [125]. In this study, long-term gastrointestinal colonization of antibiotic-treated mice by C. albicans was coupled with serial fecal transplants from colonized to naï;ve hosts. After 8-10 weekly serial passages it was verified a significantly increased intra-gastrointestinal competitive fitness, at similar levels to strains deficient in filamentous growth known to have increased fitness in antibiotic-treated mouse gut [126][127][128]. In fact, all C. albicans populations were observed to progressively lose their ability to form true hyphae when evolved in the presence of antibiotics [125].
–
Another study addressing the adaption of C. albicans isolates recovered from cystic fibrosis patients also pointed to morphological changes as a possible host-associated adaption mechanism [129]. In this case, several C. albicans isolates were seen to acquire a filamentation phenotype, underlined by mutations in the filamentation transcriptional repressor NRG1. Notably, distinct strains acquired dissimilar mutations, albeit all being located before or within the DNA binding domain of NRG1. As such, the observed filamentous growth phenotypes were associated to the likely loss of function of Nrg1 [129].
ADAPTION TO THE HUMAN IMMUNE SYSTEM
C. glabrata evolution towards increased oxidative stress resistance
–
Upon phagocytosis by the host immune system, pathogens are subject to oxidative burst [130][131]. C. glabrata shows increased tolerance to oxidative stress when compared to other yeasts, including Saccharomyces cerevisiae and C. albicans [132]. Resistance to oxidative stress and detoxification of Reactive Oxygen Species (ROS) is mainly associated with the activity of the catalase Cta1, the superoxide dismutases Sod1/Sod2 and the glutathione and thioredoxin pathways [132][133][134][135]. As shown by several genome-wide studies, regulation of the oxidative stress response is governed by the transcription factor Yap1 in vitro [136][137][138][139], however, Yap1 does not appear to be a virulence determinant and works in conjunction with other oxidative stress resistance (OSR) determinants to mediate phagocyte survival [137][140]. The ability of C. glabrata to overcome oxidative stress was inspected by in vitro evolution during oxidative (H2O2) challenge [141]. Individual C. glabrata populations were serially passaged in the presence of increasing concentrations of H2O2, resulting in a significant increase of MIC50 of H2O2 in comparison to the initial population. Increased tolerance and resistance to H2O2 is a result of adaptive evolution to oxidative stress, based on increased survival frequency of the evolved populations. Evolved strains were seen to adapt faster to hydrogen peroxide stress. In addition, adapted strains were capable of faster H2O2 detoxification and growth in higher concentrations of hydrogen peroxide than the parental strains [141]. The study resorted to whole genome sequencing and transcriptomics analysis of adapted mutants to identify the molecular basis for the evolution of populations to oxidative stress. Whole genome sequencing on selected evolved strains showed a total of 47 mutations (28 inside coding sequences). Interestingly, eleven of the mutated genes are known to affect H2O2 resistance when inactivated in S. cerevisiae. Among them, is a homolog of the MGA2 transcription factor (CAGL0F06831g), a regulator of lipid modulation in S. cerevisiae that confers H2O2 resistance when inactivated in this species [142]. Therefore, the occurrence of a mutation in C. glabrata MGA2, taking place in an evolved strain, possibly contributes to changes in membrane fatty acid composition that mediate H2O2 resistance. In fact, transcriptomics analysis during oxidative stress between evolved strains and their parental strains revealed that modulation of membrane composition is potentially involved in adaptation of C. glabrata to H2O2 [141]. Genes involved in amino acids and fatty acids were commonly upregulated among more than one evolved strain. Additional processes overrepresented in differentially expressed genes comprise cell wall remodeling and NADPH regeneration. Interestingly, only three genes were commonly up-regulated in at least three of the four adapted strains probed in the referred study. All of them have homologs involved in H2O2 resistance in S. cerevisiae. These genes (YDJ1, PDC1 and TAL1) could therefore represent interesting targets to study adaption of C. glabrata to oxidative stress. Moreover, a nonsense mutation in C. glabrata CTH2 was also found in an evolved strain. In both C. glabrata and S. cerevisiae, CTH2 is known for its role in the post-transcriptional regulation of iron limitation response through degradation of mRNAs encoding proteins involved in iron-dependent processes [143][144]. Nevertheless, increased expression of this gene contributes to enhanced ROS tolerance in S. cerevisiae [145]. The mutation of C. glabrata CTH2 was shown to contribute directly for H2O2 tolerance, as a reference strain containing the mutated CHT2 displayed significant more tolerance than the same strain carrying the wild type gene upon exposure to a high concentration of H2O2 [141].
–
Candida evolution during macrophage interaction
–
Interaction with phagocytes is another relevant topic to better understand how pathogens overcome the host immune system. More than just survive the harsh environment of the phagosome, virulence traits include the ability to escape engulfment by phagocytic cells or avoid immune recognition all together.
–
Despite belonging to the same genus, C. albicans and C. glabrata display distinct virulence features during macrophage interaction. One of the most striking differences is in their ability or inability to form hyphae. Formation of hyphae by C. albicans is associated with several virulence features, namely biofilm formation, host tissue invasion and macrophage escape [10][12][14][15]. On the other hand, C. glabrata is regarded to exist mainly in yeast form, forming pseudohyphae under particular conditions and not producing true hyphae [146]. Therefore, it lacks the yeast-to-hyphae dimorphism that comprises one of the main virulence factors of C. albicans [147].
–
Interestingly, a work by Brunke et al. has shown that C. glabrata cells co-incubated during 6 months with macrophages were able to produce pseudohyphae structures and evolve into a hypervirulent phenotype [148]. Daily passages of C. glabrata cells to new macrophage cultures resulted in the appearance of the first altered morphology cells after 1 month. The morphology of the evolved strain consisted of elongated clumps of several cells, resembling pseudohyphae structures. This change was associated with a wrinkled colony phenotype, was found to be genetically stable and to be correlated with changes in the thickness of the cell wall. The macrophage-evolved strain harbored a non-synonymous mutation in the catalytic region of the chitin synthase encoding CHS2 gene, which was shown to be responsible for its pseudohyphae growth morphology. Moreover, it was associated with a reduction in accessible chitin and reduced content of mannan and β-glucan. During co-incubation with macrophages, the expansion of the evolved strain was seen to increase continuously. As part of macrophage adaptation, the evolved strain presented a fitness advantage in comparison to wild type C. glabrata cells; but surprisingly, the increased fitness was not associated with increased tolerance to typical phagosome stress conditions. Another interesting finding was the specific microevolution of C. glabrata to host cell types. The evolved C. glabrata strain resulting from co-incubation with macrophages was seen to do significant more damage to macrophages than the parental strain after 24h, but the same was not observed for epithelial cells. Higher macrophage damage by the evolved strain was associated with faster escape after phagocytosis. Concordantly, the parental strain took four days to kill macrophages, possibly due to overgrowth of fungal mass; however, the evolved strain was able to burst macrophage cells after only 48h and continue to replicate in pseudohyphal form. These findings indicate that C. glabrata is able to evolve in a host cell-type specific manner. The microevolution of C. glabrata upon macrophage co-incubation also resulted in increased virulence in comparison to the parental strain, which was dependent on the point mutation in CHS2 that induced pseudohyphal growth [148].
–
A similar study by Wartenberg et al. has probed the microevolution of a cph1∆/efg1∆ C. albicans strain during macrophage interaction [149]. In this species, CPH1 and EFG1 are two of the main regulators of the hyphal regulatory network [150][151][152][153]. As a result, the double mutant strain is incapable of hyphal growth and macrophage escape [149]. Surprisingly, microevolution of the referred mutant during co-incubation with macrophages yielded the formation of filamentous structures (mixture of pseudohyphae and true hyphae) after 19 daily passages. This phenotype was also observed in response to several filamentation inducing conditions in vitro, including serum or N-acetyl-D-glucosamine as sole carbon source. These findings indicate that microevolution during macrophage coincubation potentiated new pathways for filamentation in C. albicans. The regain of filamentation capacity allowed the evolved strain to escape macrophages (from totally incapable in the original mutant), albeit not as efficiently as the wild type [149]. The same trend was verified in terms of adhesion potential and epithelia invasion. An increase in the damage of the evolved strain relative to its original mutant was yet observed for both macrophages and epithelial cells [149]. Cell wall architecture was also partially restored in the evolved strain, reverting the reduced mannan and increased β-glucan levels in the original mutant. It appears to be clear that the absence of two major filamentation regulators is too severe to regain full wild type-comparable virulence, but the fact that the evolved strain was able to recover partial virulence features underscores the importance of microevolution studies in the study of infection by fungal pathogens. Consistent with the previous observations, the evolved strain upregulates hyphal specific genes (e.g., HWP1, ALS3 and ECE1) and the hyphal morphogenesis regulator UME6 [149]. Moreover, the upregulation of SAP and cell wall biogenesis genes was observed. Altogether, the expression program of the evolved strain appears to underscore the regained ability of filamentous growth and host cell damage. Furthermore, the evolved strain harbored a non-synonymous mutation in the activation segment of the catalytic domain of the kinase SSN3 gene, which was shown to confer the ability to produce hyphae and damage macrophages after macrophage microevolution, even in the absence of Efg1 and Cph1 [149].
–
TABLE 1. Reported mechanisms of adaption to antifungal treatment, host niches and immune attack in C. albicans and C. glabrata as determined by microevolution studies. Both in vivo or in vitro studies are considered. The main features found to be associated with evolution events in each condition are displayed |
 |
CONCLUSIONS
The use of microevolution experiments has proven to be a relevant research topic to expand knowledge on how to tackle infections by Candida species. In this review, the data achieved by microevolution experiments to antifungal treatment, biofilm formation and host-associated stresses has been detailed.
–
Resistance to azoles in C. glabrata is largely credited to the occurrence of GOF mutations and resulting hyperactivity of the transcription factor Pdr1. Similarly, the easily acquired mutations in FKS genes are the most described mechanism of echinocandin acquired resistance in C. glabrata. In C. albicans, resistance to azoles develops in a stepwise manner with acquired mutations in drug resistance genes, such as TAC1, MRR1 and ERG11, followed by LOH events and additional gain of identical copies of the mutated genes. So far as we know, studies regarding C. albicans resistance evolution toward echinocandins are yet to be performed.
–
The information relative to evolution of biofilm formation is much more limited, but current data seems to show that the ability to form biofilm could be intrinsic to a given isolate, once the amount of biofilm formation does not appear to change in most cases over the course of an infection in each patient. Studies addressing the molecular basis that supports differential biofilm production across distinct isolates could help to pinpoint key mechanisms for biofilm formation and potentially the most relevant therapeutic targets. Moreover, co-infection with other species seems to influence this trait.
–
Evolution of C. albicans in the human host appears to result in genotypic and phenotypic variability, possibly as an approach to select for advantageous features. It was reported that C. albicans suffers changes in its competitive fitness, including changes in energy metabolism, reduction of growth rates and attenuated virulence, possibly as a persistence strategy. When probing evolution during immune encounter, both C. albicans and C. glabrata escape macrophage phagocytosis by producing hyphae or pseudohyphae, respectively. In both species, the production of pseudohyphae was associated with changes in cell wall and consistent with morphological changes. Moreover, the evolution to produce filaments was associated with mutations in key genes encoding: the chitin synthase Csh2 in C. glabrata and the Ssn3 kinase in C. albicans.
–
Interestingly, both C. albicans and C. glabrata appear to rely heavily on the occurrence of mutations, ultimately selecting a SNP or a set of SNPs that confer an evolutionary advantage. Also, this strategy takes place in more than just one selective pressure, indicating that is one of the preferred mechanisms for microevolution of these pathogens. The data from experimental evolution emphasizes the notion that resistance is a multifactorial process, often achieved in a stepwise fashion and through the combination of multiple mechanisms (Table1; Figures 1 and 2). Additionally, it is also possible that serial isolates from the same patient can result in resistance caused by different trajectories, which emphasizes the need to better understand evolutionary dynamics.
–
More than just identifying possible mechanisms of adaptation to most stress conditions human pathogens encounter, it is important to clinically validate the experimentally obtained clues. One good example of the occasional inconsistency between lab and clinical data is the participation of C. glabrata ERG11 gene in azole resistance. Although this was observed in laboratorial experiments, through ERG11 upregulation [154][155] or increased protein abundance [156], however, these mechanisms do not appear to play such a prominent role in clinical resistant isolates [41][157][158]. On the other hand, additional mechanisms of echinocandin resistance other than FKS mutations are starting to be identified [159], but the assessment of such cases in large isolate collections needs to be conducted in order to understand their clinical significance in comparison to FKS mutations. The full understanding of stress adaptation in human pathogens requires the integration of several sources of evidence to fully understand the most relevant resistance mechanisms, their regulation, their activation conditions and how they evolve in the clinical setting.
REFERENCES
- Pfaller MA, and Diekema DJ (2010). Epidemiology of invasive mycoses in North America. Crit Rev Microbiol 36(1): 1–53. doi: 10.3109/10408410903241444
- Azie N, Neofytos D, Pfaller M, Meier-Kriesche H-U, Quan S-P, and Horn D (2012). The PATH (Prospective Antifungal Therapy) Alliance® registry and invasive fungal infections: update 2012. Diagn Microbiol Infect Dis 73(4): 293–300. doi: 10.1016/j.diagmicrobio.2012.06.012
- Wisplinghoff H, Bischoff T, Tallent SM, Seifert H, Wenzel RP, and Edmond MB (2004). Nosocomial bloodstream infections in US hospitals: analysis of 24,179 cases from a prospective nationwide surveillance study. Clin Infect Dis 39(3): 309–17. doi: 10.1086/421946
- Edmond MB, Wallace SE, McClish DK, Pfaller MA, Jones RN, and Wenzel RP (1999). Nosocomial bloodstream infections in United States hospitals: a three-year analysis. Clin Infect Dis 29(2): 239–44. doi: 10.1086/520192
- Khatib R, Johnson LB, Fakih MG, Riederer K, and Briski L (2016). Current trends in candidemia and species distribution among adults: Candida glabrata surpasses C. albicans in diabetic patients and abdominal sources. Mycoses 59(12): 781–786. doi: 10.1111/myc.12531
- Tsai H-F, Krol AA, Sarti KE, and Bennett JE (2006). Candida glabrata PDR1, a transcriptional regulator of a pleiotropic drug resistance network, mediates azole resistance in clinical isolates and petite mutants. Antimicrob Agents Chemother 50(4): 1384–92. doi: 10.1128/AAC.50.4.1384-1392.2006
- Vermitsky J-P, Earhart KD, Smith WL, Homayouni R, Edlind TD, and Rogers PD (2006). Pdr1 regulates multidrug resistance in Candida glabrata: gene disruption and genome-wide expression studies. Mol Microbiol 61(3): 704–22. doi: 10.1111/j.1365-2958.2006.05235.x
- Caudle KE, Barker KS, Wiederhold NP, Xu L, Homayouni R, and Rogers PD (2011). Genomewide expression profile analysis of the Candida glabrata Pdr1 regulon. Eukaryot Cell 10(3): 373–83. doi: 10.1128/EC.00073-10
- Whaley SG, and Rogers PD (2016). Azole Resistance in Candida glabrata. Curr Infect Dis Rep 18(12): 41. doi: 10.1007/s11908-016-0554-5
- Cavalheiro M, Costa C, Pais P, and Teixeira MC (2017). Acquition of Resistance to Azole Antifungal Drugs in Candida Species: Imidazoles versus Triazoles. In: Adv Med Biol, pp 183–210.
- Cavalheiro M, and Teixeira MC (2018). Candida Biofilms: Threats, Challenges, and Promising Strategies. Front Med 5: 28. doi: 10.3389/fmed.2018.00028
- Brunke S, and Hube B (2013). Two unlike cousins: Candida albicans and C. glabrata infection strategies. Cell Microbiol 15(5): 701-8. doi: 10.1111/cmi.12091
- Kasper L, Seider K, and Hube B (2015). Intracellular survival of Candida glabrata in macrophages: Immune evasion and persistence. FEMS Yeast Res 15(5): fov042. doi: 10.1093/femsyr/fov042
- Uwamahoro N, Verma-Gaur J, Shen HH, Qu Y, Lewis R, Lu J, Bambery K, Masters SL, Vince JE, Naderer T, and Traven A (2014). The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. MBio 5(2): e00003-14. doi: 10.1128/mBio.00003-14
- Lorenz MC, Bender JA, and Fink GR (2004). Transcriptional response of Candida albicans upon internalization by macrophages. Eukaryot Cell 3(5): 1076-87. doi: 10.1128/EC.3.5.1076-1087.2004
- Li L, Kashleva H, and Dongari-Bagtzoglou A (2007). Cytotoxic and cytokine-inducing properties of Candida glabrata in single and mixed oral infection models. Microb Pathog 42(4): 138–47. doi: 10.1016/j.micpath.2006.12.003
- Seider K, Brunke S, Schild L, Jablonowski N, Wilson D, Majer O, Barz D, Haas A, Kuchler K, Schaller M, and Hube B (2011). The facultative intracellular pathogen Candida glabrata subverts macrophage cytokine production and phagolysosome maturation. J Immunol 187(6): 3072–86. doi: 10.4049/jimmunol.1003730
- Dementhon K, El-Kirat-Chatel S, and Noël T (2012). Development of an in vitro model for the Multi-Parametric quantification of the cellular interactions between Candida yeasts and phagocytes. PLoS One 7(3): e32621. doi: 10.1371/journal.pone.0032621
- Naglik J, Albrecht A, Bader O, and Hube B (2004). Candida albicans proteinases and host/pathogen interactions. Cell Microbiol 6(10): 915-26. doi: 10.1111/j.1462-5822.2004.00439.x
- Kantarcioglu AS, and Yücel A (2002). Phospholipase and protease activities in clinical Candida isolates with reference to the sources of strains. Mycoses 45(5–6): 160–5. doi: 10.1046/j.1439-0507.2002.00727.x
- Kaur R, Ma B, and Cormack BP (2007). A family of glycosylphosphatidylinositol-linked aspartyl proteases is required for virulence of Candida glabrata. Proc Natl Acad Sci 104(18): 7628-33. doi: 10.1073/pnas.0611195104
- Petrikkos G, and Skiada A (2007). Recent advances in antifungal chemotherapy. Int J Antimicrob Agents 30(2): 108–17. doi: 10.1016/j.ijantimicag.2007.03.009
- Ostrosky-Zeichner L, Casadevall A, Galgiani JN, Odds FC, and Rex JH (2010). An insight into the antifungal pipeline: selected new molecules and beyond. Nat Rev Drug Discov 9(9): 719–727. doi: 10.1038/nrd3074
- Borst A, Raimer MT, Warnock DW, Morrison CJ, and Arthington- Skaggs BA (2005). Rapid acquisition of stable azole resistance by Candida glabrata isolates obtained before the clinical introduction of fluconazole. Antimicrob Agents Chemother 49(2): 783–7. doi: 10.1128/AAC.49.2.783-787.2005
- Wiederhold N (2017). Antifungal resistance: current trends and future strategies to combat. Infect Drug Resist 10: 249–259. doi: 10.2147/IDR.S124918
- Shapiro RS, Robbins N, and Cowen LE (2011). Regulatory circuitry governing fungal development, drug resistance, and disease. Microbiol Mol Biol Rev 75(2): 213–67. doi: 10.1128/MMBR.00045-10
- Anderson JB (2005). Evolution of antifungal-drug resistance: mechanisms and pathogen fitness. Nat Rev Microbiol 3(7): 547–556. doi: 10.1038/nrmicro1179
- Mukherjee PK, Chandra J, Kuhn DM, and Ghannoum MA (2003). Mechanism of fluconazole resistance in Candida albicans biofilms: phase-specific role of efflux pumps and membrane sterols. Infect Immun 71(8): 4333–40. doi: 10.1128/IAI.71.8.4333-4340.2003
- MacPherson S, Akache B, Weber S, De Deken X, Raymond M, and Turcotte B (2005). Candida albicans Zinc Cluster Protein Upc2p Confers Resistance to Antifungal Drugs and Is an Activator of Ergosterol Biosynthetic Genes. Antimicrob Agents Chemother 49(5): 1745–1752. doi: 10.1128/AAC.49.5.1745-1752.2005
- Cowen LE, and Steinbach WJ (2008). Stress, drugs, and evolution: the role of cellular signaling in fungal drug resistance. Eukaryot Cell 7(5): 747–64. doi: 10.1128/EC.00041-08
- Hoot SJ, Smith AR, Brown RP, and White TC (2011). An A643V amino acid substitution in Upc2p contributes to azole resistance in well-characterized clinical isolates of Candida albicans. Antimicrob Agents Chemother 55(2): 940–2. doi: 10.1128/AAC.00995-10
- Heilmann CJ, Schneider S, Barker KS, Rogers PD, and Morschhäuser J (2010). An A643T mutation in the transcription factor Upc2p causes constitutive ERG11 upregulation and increased fluconazole resistance in Candida albicans. Antimicrob Agents Chemother 54(1): 353–9. doi: 10.1128/AAC.01102-09
- Flowers SA, Barker KS, Berkow EL, Toner G, Chadwick SG, Gygax SE, Morschhäuser J, and Rogers PD (2012). Gain-of-function mutations in UPC2 are a frequent cause of ERG11 upregulation in azole-resistant clinical isolates of Candida albicans. Eukaryot Cell 11(10): 1289–99. doi: 10.1128/EC.00215-12
- Dunkel N, Blaß J, Rogers PD, and Morschhäuser J (2008). Mutations in the multi-drug resistance regulator MRR1, followed by loss of heterozygosity, are the main cause of MDR1 overexpression in fluconazole-resistant Candida albicans strains. Mol Microbiol 69(4): 827–840. doi: 10.1111/j.1365-2958.2008.06309.x
- Coste A, Turner V, Ischer F, Morschhäuser J, Forche A, Selmecki A, Berman J, Bille J, and Sanglard D (2006). A mutation in Tac1p, a transcription factor regulating CDR1 and CDR2, is coupled with loss of heterozygosity at chromosome 5 to mediate antifungal resistance in Candida albicans. Genetics 172(4): 2139–2156. doi: 10.1534/genetics.105.054767
- Dunkel N, Liu TT, Barker KS, Homayouni R, Morschhäuser J, and Rogers PD (2008). A gain-of-function mutation in the transcription factor Upc2p causes upregulation of ergosterol biosynthesis genes and increased fluconazole resistance in a clinical Candida albicans isolate. Eukaryot Cell 7(7): 1180–90. doi: 10.1128/EC.00103-08
- Selmecki A, Forche A, and Berman J (2006). Aneuploidy and Isochromosome Formation in Drug-Resistant Candida albicans. Science 313(5785): 367–370. doi: 10.1126/science.1128242
- Selmecki A, Gerami-Nejad M, Paulson C, Forche A, and Berman J (2008). An isochromosome confers drug resistance in vivo by amplification of two genes, ERG11 and TAC1. Mol Microbiol 68(3): 624–641. doi: 10.1111/j.1365-2958.2008.06176.x
- Selmecki AM, Dulmage K, Cowen LE, Anderson JB, and Berman J (2009). Acquisition of Aneuploidy Provides Increased Fitness during the Evolution of Antifungal Drug Resistance. PLoS Genet 5(10): e1000705. doi: 10.1371/journal.pgen.1000705
- Gao J, Wang H, Li Z, Wong AH-H, Wang Y-Z, Guo Y, Lin X, Zeng G, Wang Y, and Wang J (2018). Candida albicans gains azole resistance by altering sphingolipid composition. Nat Commun 9(1): 4495. doi: 10.1038/s41467-018-06944-1
- Vermitsky J-P, and Edlind TD (2004). Azole resistance in Candida glabrata: coordinate upregulation of multidrug transporters and evidence for a Pdr1-like transcription factor. Antimicrob Agents Chemother 48(10): 3773–81. doi: 10.1128/AAC.48.10.3773-3781.2004
- Sanguinetti M, Posteraro B, Fiori B, Ranno S, Torelli R, and Fadda G (2005). Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob Agents Chemother 49(2): 668–79. doi: 10.1128/AAC.49.2.668-679.2005
- vanden Bossche H, Marichal P, Odds FC, Le Jeune L, and Coene MC (1992). Characterization of an azole-resistant Candida glabrata isolate. Antimicrob Agents Chemother 36(12): 2602–10. doi: 10.1128/AAC.36.12.2602
- Sanglard D, Ischer F, Calabrese D, Majcherczyk PA, and Bille J (1999). The ATP binding cassette transporter gene CgCDR1 from Candida glabrata is involved in the resistance of clinical isolates to azole antifungal agents. Antimicrob Agents Chemother 43(11): 2753–65. doi: 10.1128/aac.43.11.2753
- Costa C, Nunes J, Henriques A, Mira NP, Nakayama H, Chibana H, and Teixeira MC (2014). Candida glabrata drug:H+ antiporter CgTpo3 (ORF CAGL0I10384g): role in azole drug resistance and polyamine homeostasis. J Antimicrob Chemother 69(7): 1767–1776. doi: 10.1093/jac/dku044
- Costa C, Pires C, Cabrito TR, Renaudin A, Ohno M, Chibana H, Sá- Correia I, and Teixeira MC (2013). Candida glabrata drug: H+antiporter CgQdr2 confers imidazole drug resistance, being activated by transcription factor CgPdr1. Antimicrob Agents Chemother 57(7): 3159-67. doi: 10.1128/AAC.00811-12
- Sanglard D, Ischer F, and Bille J (2001). Role of ATP-binding- cassette transporter genes in high-frequency acquisition of resistance to azole antifungals in Candida glabrata. Antimicrob Agents Chemother 45(4): 1174–83. doi: 10.1128/AAC.45.4.1174-1183.2001
- Ferrari S, Ischer F, Calabrese D, Posteraro B, Sanguinetti M, Fadda G, Rohde B, Bauser C, Bader O, and Sanglard D (2009). Gain of Function Mutations in CgPDR1 of Candida glabrata Not Only Mediate Antifungal Resistance but Also Enhance Virulence. PLoS Pathog 5(1): e1000268. doi: 10.1371/journal.ppat.1000268
- Defontaine A, Bouchara J-P, Declerk P, Planchenault C, Chabasse D, and Hallet J-N (1999). In-vitro resistance to azoles associated with mitochondrial DNA deficiency in Candida glabrata. J Med Microbiol 48(7): 663–670. doi: 10.1099/00222615-48-7-663
- Paul S, Schmidt JA, and Moye-Rowley WS (2011). Regulation of the CgPdr1 Transcription Factor from the Pathogen Candida glabrata. Eukaryot Cell 10(2): 187–197. doi: 10.1128/EC.00277-10
- Kaur R, Castaño I, and Cormack BP (2004). Functional genomic analysis of fluconazole susceptibility in the pathogenic yeast Candida glabrata: roles of calcium signaling and mitochondria. Antimicrob Agents Chemother 48(5): 1600–13. doi: 10.1128/AAC.48.5.1600-1613.2004
- Ferrari S, Sanguinetti M, De Bernardis F, Torelli R, Posteraro B, Vandeputte P, and Sanglard D (2011). Loss of mitochondrial functions associated with azole resistance in Candida glabrata results in enhanced virulence in mice. Antimicrob Agents Chemother 55(5): 1852–60. doi: 10.1128/AAC.01271-10
- Salazar SB, Wang C, Münsterkötter M, Okamoto M, Takahashi- Nakaguchi A, Chibana H, Lopes MM, Güldener U, Butler G, and Mira NP (2018). Comparative genomic and transcriptomic analyses unveil novel features of azole resistance and adaptation to the human host in Candida glabrata. FEMS Yeast Res 18(1). doi: 10.1093/femsyr/fox079
- Denning DW (2003). Echinocandin antifungal drugs. Lancet 362(9390): 1142–1151. doi: 10.1016/S0140-6736(03)14472-8
- Bachmann SP, VandeWalle K, Ramage G, Patterson TF, Wickes BL, Graybill JR, and López-Ribot JL (2002). In vitro activity of caspofungin against Candida albicans biofilms. Antimicrob Agents Chemother 46(11): 3591–6. doi: 10.1128/AAC.46.11.3591-3596.2002
- Kuhn DM, George T, Chandra J, Mukherjee PK, and Ghannoum MA (2002). Antifungal susceptibility of Candida biofilms: unique efficacy of amphotericin B lipid formulations and echinocandins. Antimicrob Agents Chemother 46(6): 1773–80. doi: 10.1128/AAC.46.6.1773-1780.2002
- Sanguinetti M, Posteraro B, and Lass-Flörl C (2015). Antifungal drug resistance among Candida species: mechanisms and clinical impact. Mycoses 58: 2–13. doi: 10.1111/myc.12330
- Cornely OA, Bassetti M, Calandra T, Garbino J, Kullberg BJ, Lortholary O, Meersseman W, Akova M, Arendrup MC, Arikan-Akdagli S, Bille J, Castagnola E, Cuenca-Estrella M, Donnelly JP, Groll AH, Herbrecht R, Hope WW, Jensen HE, Lass-Florl C, Petrikkos G, Richardson MD, Roilides E, Verweij PE, Viscoli C, Ullmann AJ, and ESCMID Fungal Infection Study Group (2012). ESCMID* *This guideline was presented in part at ECCMID 2011. European Society for Clinical Microbiology and Infectious Diseases. guideline for the diagnosis and management of Candida diseases 2012: non-neutropenic adult patients. Clin Microbiol Infect 18: 19–37. doi: 10.1111/1469-0691.12039
- Katiyar S, Pfaller M, and Edlind T (2006). Candida albicans and Candida glabrata clinical isolates exhibiting reduced echinocandin susceptibility. Antimicrob Agents Chemother 50(8): 2892–4. doi: 10.1128/AAC.00349-06
- Park S, Kelly R, Kahn JN, Robles J, Hsu M-J, Register E, Li W, Vyas V, Fan H, Abruzzo G, Flattery A, Gill C, Chrebet G, Parent SA, Kurtz M, Teppler H, Douglas CM, and Perlin DS (2005). Specific substitutions in the echinocandin target Fks1p account for reduced susceptibility of rare laboratory and clinical Candida sp. isolates. Antimicrob Agents Chemother 49(8): 3264–73. doi: 10.1128/AAC.49.8.3264-3273.2005
- Costa-de-Oliveira S, Marcos Miranda I, Silva RM, Pinto E Silva A, Rocha R, Amorim A, Gonçalves Rodrigues A, and Pina-Vaz C (2011). FKS2 mutations associated with decreased echinocandin susceptibility of Candida glabrata following anidulafungin therapy. Antimicrob Agents Chemother 55(3): 1312–4. doi: 10.1128/AAC.00589-10
- Douglas CM, D’Ippolito JA, Shei GJ, Meinz M, Onishi J, Marrinan JA, Li W, Abruzzo GK, Flattery A, Bartizal K, Mitchell A, and Kurtz MB (1997). Identification of the FKS1 gene of Candida albicans as the essential target of 1,3-beta-D-glucan synthase inhibitors. Antimicrob Agents Chemother 41(11): 2471–9. doi: 10.1128/AAC.41.11.2471
- Katiyar SK, Alastruey-Izquierdo A, Healey KR, Johnson ME, Perlin DS, and Edlind TD (2012). Fks1 and Fks2 are functionally redundant but differentially regulated in Candida glabrata: implications for echinocandin resistance. Antimicrob Agents Chemother 56(12): 6304–9. doi: 10.1128/AAC.00813-12
- Dannaoui E, Desnos-Ollivier M, Garcia-Hermoso D, Grenouillet F, Cassaing S, Baixench M-T, Bretagne S, Dromer F, Lortholary O, and French Mycoses Study Group the FMS (2012). Candida spp. with acquired echinocandin resistance, France, 2004-2010. Emerg Infect Dis 18(1): 86–90. doi: 10.3201/eid1801.110556
- Matsumoto E, Boyken L, Tendolkar S, McDanel J, Castanheira M, Pfaller M, and Diekema D (2014). Candidemia surveillance in Iowa: emergence of echinocandin resistance. Diagn Microbiol Infect Dis 79(2): 205–8. doi: 10.1016/j.diagmicrobio.2014.02.016
- Gray KC, Palacios DS, Dailey I, Endo MM, Uno BE, Wilcock BC, and Burke MD (2012). Amphotericin primarily kills yeast by simply binding ergosterol. Proc Natl Acad Sci U S A 109(7): 2234–9. doi: 10.1073/pnas.1117280109
- Lemke A, Kiderlen AF, and Kayser O (2005). Amphotericin B. Appl Microbiol Biotechnol 68(2): 151–162. doi: 10.1007/s00253-005-1955-9
- Cho E-J, Shin JH, Kim SH, Kim H-K, Park JS, Sung H, Kim M-N, and Im HJ (2014). Emergence of multiple resistance profiles involving azoles, echinocandins and amphotericin B in Candida glabrata isolates from a neutropenia patient with prolonged fungaemia. J Antimicrob Chemother 70(4): 1268–1270. doi: 10.1093/jac/dku518
- Krogh-Madsen M, Arendrup MC, Heslet L, and Knudsen JD (2006). Amphotericin B and Caspofungin Resistance in Candida glabrata Isolates Recovered from a Critically Ill Patient. Clin Infect Dis 42(7): 938–944. doi: 10.1086/500939
- Martel CM, Parker JE, Bader O, Weig M, Gross U, Warrilow AGS, Kelly DE, and Kelly SL (2010). A Clinical Isolate of Candida albicans with Mutations in ERG11 (Encoding Sterol 14 -Demethylase) and ERG5 (Encoding C22 Desaturase) Is Cross Resistant to Azoles and Amphotericin B. Antimicrob Agents Chemother 54(9): 3578–3583. doi: 10.1128/AAC.00303-10
- Vandeputte P, Tronchin G, Larcher G, Ernoult E, Bergès T, Chabasse D, and Bouchara J-P (2008). A nonsense mutation in the ERG6 gene leads to reduced susceptibility to polyenes in a clinical isolate of Candida glabrata. Antimicrob Agents Chemother 52(10): 3701–9. doi: 10.1128/AAC.00423-08
- Robbins N, Wright GD, and Cowen LE (2016). Antifungal Drugs: The Current Armamentarium and Development of New Agents. In: The Fungal Kingdom. American Society of Microbiology 4(5): 903–922. doi: 10.1128/microbiolspec.FUNK-0002-2016
- Vandeputte P, Pineau L, Larcher G, Noel T, Brèthes D, Chabasse D, and Bouchara J-P (2011). Molecular Mechanisms of Resistance to 5- Fluorocytosine in Laboratory Mutants of Candida glabrata. Mycopathologia 171(1): 11–21. doi: 10.1007/s11046-010-9342-1
- Hope WW, Tabernero L, Denning DW, and Anderson MJ (2004). Molecular Mechanisms of Primary Resistance to Flucytosine in Candida albicans. Antimicrob Agents Chemother 48(11): 4377–4386. doi: 10.1128/AAC.48.11.4377-4386.2004
- Costa C, Ponte A, Pais P, Santos R, Cavalheiro M, Yaguchi T, Chibana H, and Teixeira MC (2015). New mechanisms of flucytosine resistance in C. glabrata unveiled by a chemogenomics analysis in S. cerevisiae. PLoS One 10(8): e0135110. doi: 10.1371/journal.pone.0135110
- Barrick JE, and Lenski RE (2013). Genome dynamics during experimental evolution. Nat Rev Genet 14(12): 827–839. doi: 10.1038/nrg3564
- Elena SF, and Lenski RE (2003). Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat Rev Genet 4(6): 457–469. doi: 10.1038/nrg1088
- Andes D, Forrest A, Lepak A, Nett J, Marchillo K, and Lincoln L (2006). Impact of Antimicrobial Dosing Regimen on Evolution of Drug Resistance In Vivo: Fluconazole and Candida albicans. Antimicrob Agents Chemother 50(7): 2374–2383. doi: 10.1128/AAC.01053-05
- Ben-Ami R, Zimmerman O, Finn T, Amit S, Novikov A, Wertheimer N, Lurie-Weinberger M, and Berman J (2016). Heteroresistance to Fluconazole Is a Continuously Distributed Phenotype among Candida glabrata Clinical Strains Associated with In Vivo Persistence. MBio 7(4): e00655-16. doi: 10.1128/mBio.00655-16
- Tian Y, Gao N, Ni Q, Mao Y, Dong D, Huang X, Jiang C, Li Z, Zhang L, Wang X, Peng Y, and Chen C (2018). Sequence modification of the master regulator Pdr1 interferes with its transcriptional autoregulation and confers altered azole resistance in Candida glabrata. FEMS Yeast Res 18(4): foy038. doi: 10.1093/femsyr/foy038
- Hou X, Xiao M, Wang H, Yu S-Y, Zhang G, Zhao Y, and Xu Y-C (2018). Profiling of PDR1 and MSH2 in Candida glabrata Bloodstream Isolates from a Multi-Center Study in China. Antimicrob Agents Chemother 62(6): e00153-18. doi: 10.1128/AAC.00153-18
- Katiyar S, Shiffrin E, Shelton C, Healey K, Vermitsky JP, and Edlind T (2016). Evaluation of polymorphic locus sequence typing for candida glabrata epidemiology. J Clin Microbiol 54(4): 1042-50. doi: 10.1128/JCM.03106-15
- Cowen LE, Sanglard D, Calabrese D, Sirjusingh C, Anderson JB, and Kohn LM (2000). Evolution of drug resistance in experimental populations of Candida albicans. J Bacteriol 182(6): 1515–22. doi: 10.1128/jb.182.6.1515-1522.2000
- Vincent BM, Lancaster AK, Scherz-Shouval R, Whitesell L, and Lindquist S (2013). Fitness Trade-offs Restrict the Evolution of Resistance to Amphotericin B. PLoS Biol 11(10): e1001692. doi: 10.1371/journal.pbio.1001692
- Cavalheiro M, Costa C, Silva-Dias A, Miranda IM, Wang C, Pais P, Pinto SN, Mil-Homens D, Sato-Okamoto M, Takahashi-Nakaguchi A, Silva RM, Mira NP, Fialho AM, Chibana H, Rodrigues AG, Butler G, and Teixeira MC (2019). A Transcriptomics Approach To Unveiling the Mechanisms of In Vitro Evolution towards Fluconazole Resistance of a Candida glabrata Clinical Isolate. Antimicrob Agents Chemother 63(1): e00995-18. doi: 10.1128/AAC.00995-18
- Vale-Silva L, Beaudoing E, Tran VDT, and Sanglard D (2017). Comparative Genomics of Two Sequential Candida glabrata Clinical Isolates. G3 Genes, Genomes, Genet 7(8): 2413–2426. doi: 10.1534/G3.117.042887
- Ni Q, Wang C, Tian Y, Dong D, Jiang C, Mao E, and Peng Y (2018). CgPDR1 gain-of-function mutations lead to azole-resistance and increased adhesion in clinical Candida glabrata strains. Mycoses 61(7): 430–440. doi: 10.1111/myc.12756
- Bordallo-Cardona MÁ, Escribano P, Marcos-Zambrano LJ, Díaz- García J, de la Pedrosa EG, Cantón R, Bouza E, and Guinea J (2018). Low and constant micafungin concentrations may be sufficient to lead to resistance mutations in FKS2 gene of Candida glabrata. Med Mycol 56(7): 903–906. doi: 10.1093/mmy/myx124
- Huang M, McClellan M, Berman J, and Kao KC (2011). Evolutionary Dynamics of Candida albicans during In Vitro Evolution. Eukaryot Cell 10(11): 1413–1421. doi: 10.1128/EC.05168-11
- Kao KC, and Sherlock G (2008). Molecular characterization of clonal interference during adaptive evolution in asexual populations of Saccharomyces cerevisiae. Nat Genet 40(12): 1499–1504. doi: 10.1038/ng.280
- White TC (1997). Increased mRNA levels of ERG16, CDR, and MDR1 correlate with increases in azole resistance in Candida albicans isolates from a patient infected with human immunodeficiency virus. Antimicrob Agents Chemother 41(7): 1482–7. doi: 10.1128/aac.41.7.1482
- White TC (1997). The presence of an R467K amino acid substitution and loss of allelic variation correlate with an azole- resistant lanosterol 14alpha demethylase in Candida albicans. Antimicrob Agents Chemother 41(7): 1488–94. doi: 10.1128/aac.41.7.1488
- White TC, Pfaller MA, Rinaldi MG, Smith J, and Redding SW (1997). Stable azole drug resistance associated with a substrain of Candida albicans from an HIV-infected patient. Oral Dis 3 (Suppl 1): S102-9. doi: 10.1111/j.1601-0825.1997.tb00336.x
- Coste A, Selmecki A, Forche A, Diogo D, Bougnoux ME, D’Enfert C, Berman J, and Sanglard D (2007). Genotypic evolution of azole resistance mechanisms in sequential Candida albicans isolates. Eukaryot Cell 6(10): 1889–1904. doi: 10.1128/EC.00151-07
- Popp C, Hampe IAI, Hertlein T, Ohlsen K, Rogers PD, and Morschhäuser J (2017). Competitive Fitness of Fluconazole-Resistant Clinical Candida albicans Strains. Antimicrob Agents Chemother 61(7): e00584-17. doi: 10.1128/AAC.00584-17
- Cowen LE, and Lindquist S (2005). Hsp90 Potentiates the Rapid Evolution of New Traits: Drug Resistance in Diverse Fungi. Science 309(5744): 2185–2189. doi: 10.1126/science.1118370
- Sidera K, and Patsavoudi E (2014). HSP90 inhibitors: current development and potential in cancer therapy. Recent Pat Anticancer Drug Discov 9(1): 1–20. doi: 10.2174/15748928113089990031
- Franz R, Kelly SL, Lamb DC, Kelly DE, Ruhnke M, and Morschhäuser J (1998). Multiple molecular mechanisms contribute to a stepwise development of fluconazole resistance in clinical Candida albicans strains. Antimicrob Agents Chemother 42(12): 3065–72. doi: 10.1128/aac.42.12.3065
- Morschhäuser J, Barker KS, Liu TT, Blaß-Warmuth J, Homayouni R, and Rogers PD (2007). The Transcription Factor Mrr1p Controls Expression of the MDR1 Efflux Pump and Mediates Multidrug Resistance in Candida albicans. PLoS Pathog 3(11): e164. doi: 10.1371/journal.ppat.0030164
- Ford CB, Funt JM, Abbey D, Issi L, Guiducci C, Martinez DA, Delorey T, Li B yu, White TC, Cuomo C, Rao RP, Berman J, Thompson DA, and Regev A (2015). The evolution of drug resistance in clinical isolates of Candida albicans. Elife 4: e00662. doi: 10.7554/eLife.00662
- Healey KR, Zhao Y, Perez WB, Lockhart SR, Sobel JD, Farmakiotis D, Kontoyiannis DP, Sanglard D, Taj-Aldeen SJ, Alexander BD, Jimenez- Ortigosa C, Shor E, and Perlin DS (2016). Prevalent mutator genotype identified in fungal pathogen Candida glabrata promotes multi-drug resistance. Nat Commun 7(1): 11128. doi: 10.1038/ncomms11128
- Ferrari S, Sanguinetti M, Torelli R, Posteraro B, and Sanglard D (2011). Contribution of CgPDR1-Regulated Genes in Enhanced Virulence of Azole-Resistant Candida glabrata. PLoS One 6(3): e17589. doi: 10.1371/journal.pone.0017589
- Singh-Babak SD, Babak T, Diezmann S, Hill JA, Xie JL, Chen Y-L, Poutanen SM, Rennie RP, Heitman J, and Cowen LE (2012). Global Analysis of the Evolution and Mechanism of Echinocandin Resistance in Candida glabrata. PLoS Pathog 8(5): e1002718. doi: 10.1371/journal.ppat.1002718
- Singh SD, Robbins N, Zaas AK, Schell WA, Perfect JR, and Cowen LE (2009). Hsp90 Governs Echinocandin Resistance in the Pathogenic Yeast Candida albicans via Calcineurin. PLoS Pathog 5(7): e1000532. doi: 10.1371/journal.ppat.1000532
- Cowen LE, Singh SD, Köhler JR, Collins C, Zaas AK, Schell WA, Aziz H, Mylonakis E, Perfect JR, Whitesell L, and Lindquist S (2009). Harnessing Hsp90 function as a powerful, broadly effective therapeutic strategy for fungal infectious disease. Proc Natl Acad Sci 106(8): 2818–2823. doi: 10.1073/pnas.0813394106
- Biswas C, Chen SC-A, Halliday C, Kennedy K, Playford EG, Marriott DJ, Slavin MA, Sorrell TC, and Sintchenko V (2017). Identification of genetic markers of resistance to echinocandins, azoles and 5- fluorocytosine in Candida glabrata by next-generation sequencing: a feasibility study. Clin Microbiol Infect 23(9): 676.e7-676.e10. doi: 10.1016/j.cmi.2017.03.014
- Chong PP, Chin VK, Wong WF, Madhavan P, Yong VC, and Looi CY (2018). Transcriptomic and Genomic Approaches for Unravelling Candida albicans Biofilm Formation and Drug Resistance-An Update. Genes 9(11): E540. doi: 10.3390/genes9110540
- Nobile CJ, Fox EP, Nett JE, Sorrells TR, Mitrovich QM, Hernday AD, Tuch BB, Andes DR, and Johnson AD (2012). A Recently Evolved Transcriptional Network Controls Biofilm Development in Candida albicans. Cell 148(1–2): 126–138. doi: 10.1016/j.cell.2011.10.048
- Murillo LA, Newport G, Lan C-Y, Habelitz S, Dungan J, and Agabian NM (2005). Genome-wide transcription profiling of the early phase of biofilm formation by Candida albicans. Eukaryot Cell 4(9): 1562–73. doi: 10.1128/EC.4.9.1562-1573.2005
- Chandra J, Mukherjee PK, Leidich SD, Faddoul FF, Hoyer LL, Douglas LJ, and Ghannoum MA (2001). Antifungal resistance of candidal biofilms formed on denture acrylic in vitro. J Dent Res 80(3): 903–908. doi: 10.1177/00220345010800031101
- Hawser SP, and Douglas LJ (1995). Resistance of Candida albicans biofilms to antifungal agents in vitro. Antimicrob Agents Chemother 39(9): 2128–2131. doi: 10.1128/AAC.39.9.2128
- Cavalheiro M, and Teixeira MC (2018). Candida Biofilms: Threats, Challenges, and Promising Strategies. Front Med 5(28): 1–15. doi: 10.3389/fmed.2018.00028
- Chandra J, and Mukherjee PK (2015). Candida Biofilms: Development, Architecture, and Resistance. Microbiol Spectr 3(4). doi: 10.1128/microbiolspec.MB-0020-2015
- Lohse MB, Gulati M, Johnson AD, and Nobile CJ (2018). Development and regulation of single-and multi-species Candida albicans biofilms. Nat Rev Microbiol 16(1): 19–31. doi: 10.1038/nrmicro.2017.107
- Nett J, Lincoln L, Marchillo K, Massey R, Holoyda K, Hoff B, VanHandel M, and Andes D (2007). Putative role of β-1,3 glucans in Candida albicans biofilm resistance. Antimicrob Agents Chemother 51(2): 510–520. doi: 10.1128/AAC.01056-06
- Jain N, Kohli R, Cook E, Gialanella P, Chang T, and Fries BC (2007). Biofilm Formation by and Antifungal Susceptibility of Candida Isolates from Urine. Appl Environ Microbiol 73(6): 1697–1703. doi: 10.1128/AEM.02439-06
- Hasan F, Xess I, Wang X, Jain N, and Fries BC Biofilm formation in clinical Candida isolates and its association with virulence. Microbes Infect 11(8–9): 753–61. doi: 10.1016/j.micinf.2009.04.018
- Bitar I, Khalaf RA, Harastani H, and Tokajian S (2014). Identification, typing, antifungal resistance profile, and biofilm formation of Candida albicans isolates from Lebanese hospital patients. Biomed Res Int 2014: 931372. doi: 10.1155/2014/931372
- Rafaluk C, Jansen G, Schulenburg H, and Joop G (2015). When experimental selection for virulence leads to loss of virulence. Trends Parasitol 31(9): 426-34. doi: 10.1016/j.pt.2015.06.002
- Ebert D (1998). Experimental Evolution of Parasites. Science 282(5393): 1432-36. doi: 10.1126/science.282.5393.1432
- Cheng S, Clancy CJ, Zhang Z, Hao B, Wang W, Iczkowski KA, Pfaller MA, and Nguyen MH (2007). Uncoupling of oxidative phosphorylation enables Candida albicans to resist killing by phagocytes and persist in tissue. Cell Microbiol 9(2): 492-501. doi: 10.1111/j.1462-5822.2006.00805.x
- Forche A, Magee PT, Selmecki A, Berman J, and May G (2009). Evolution in Candida albicans populations during a single passage through a mouse host. Genetics 182(3): 799-811. doi: 10.1534/genetics.109.103325
- Lionakis MS, Lim JK, Lee CCR, and Murphy PM (2011). Organ- specific innate immune responses in a mouse model of invasive candidiasis. J Innate Immun 3(2): 180-99. doi: 10.1159/000321157
- Lüttich A, Brunke S, Hube B, and Jacobsen ID (2013). Serial Passaging of Candida albicans in Systemic Murine Infection Suggests That the Wild Type Strain SC5314 Is Well Adapted to the Murine Kidney. PLoS One 8(5): e64482. doi: 10.1371/journal.pone.0064482
- Tso GHW, Reales-Calderon JA, Tan ASM, Sem XH, Le GTT, Tan TG, Lai GC, Srinivasan KG, Yurieva M, Liao W, Poidinger M, Zolezzi F, Rancati G, and Pavelka N (2018). Experimental evolution of a fungal pathogen into a gut symbiont. Science 362(6414): 589-595. doi: 10.1126/science.aat0537
- Pierce J V., and Kumamoto CA (2012). Variation in Candida albicans EFG1 expression enables host-dependent changes in colonizing fungal populations. MBio 3(4): e00117-12. doi: 10.1128/mBio.00117-12
- Hirakawa MP, Martinez DA, Sakthikumar S, Anderson MZ, Berlin A, Gujja S, Zeng Q, Zisson E, Wang JM, Greenberg JM, Berman J, Bennett RJ, and Cuomo CA (2015). Genetic and phenotypic intra- species variation in Candida albicans. Genome Res 25(3): 413-25. doi: 10.1101/gr.174623.114
- Pande K, Chen C, and Noble SM (2013). Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat Genet 45(9): 1088-91. doi: 10.1038/ng.2710
- Kim SH, Clark ST, Surendra A, Copeland JK, Wang PW, Ammar R, Collins C, Tullis DE, Nislow C, Hwang DM, Guttman DS, and Cowen LE (2015). Global Analysis of the Fungal Microbiome in Cystic Fibrosis Patients Reveals Loss of Function of the Transcriptional Repressor Nrg1 as a Mechanism of Pathogen Adaptation. PLoS Pathog 11(11): e1005308. doi: 10.1371/journal.ppat.1005308
- Vieira O V, Botelho RJ, and Grinstein S (2002). Phagosome maturation: aging gracefully. Biochem J 366(Pt 3): 689–704. doi: 10.1042/BJ20020691
- Haas A (2007). The phagosome: Compartment with a license to kill. Traffic 8(4): 311-30. doi: 10.1111/j.1600-0854.2006.00531.x
- Cuéllar-Cruz M, Briones-Martin-del-Campo M, Cañas-Villamar I, Montalvo-Arredondo J, Riego-Ruiz L, Castaño I, and De Las Peñas A (2008). High resistance to oxidative stress in the fungal pathogen Candida glabrata is mediated by a single catalase, Cta1p, and is controlled by the transcription factors Yap1p, Skn7p, Msn2p, and Msn4p. Eukaryot Cell 7(5): 814-25. doi: 10.1128/EC.00011-08
- Mahl CD, Behling CS, Hackenhaar FS, de Carvalho e Silva MN, Putti J, Salomon TB, Alves SH, Fuentefria A, and Benfato MS (2015). Induction of ROS generation by fluconazole in Candida glabrata: activation of antioxidant enzymes and oxidative DNA damage. Diagn Microbiol Infect Dis 82(3): 203–8. doi: 10.1016/j.diagmicrobio.2015.03.019
- Fukuda Y, Tsai HF, Myers TG, and Bennett JE (2013). Transcriptional profiling of Candida glabrata during phagocytosis by neutrophils and in the infected mouse spleen. Infect Immun 81(4): 1325-33. doi: 10.1128/IAI.00851-12
- Briones-Martin-Del-Campo M, Orta-Zavalza E, Juarez-Cepeda J, Gutierrez-Escobedo G, Cañas-Villamar I, Castaño I, and De Las Peñas A (2014). The oxidative stress response of the opportunistic fungal pathogen Candida glabrata. Rev Iberoam Micol 31(1): 67-71. doi: 10.1016/j.riam.2013.09.012
- Roetzer A, Gregori C, Jennings AM, Quintin J, Ferrandon D, Butler G, Kuchler K, Ammerer G, and Schüller C (2008). Candida glabrata environmental stress response involves Saccharomyces cerevisiae Msn2/4 orthologous transcription factors. Mol Microbiol 69(3): 603- 20. doi: 10.1111/j.1365-2958.2008.06301.x
- Roetzer A, Klopf E, Gratz N, Marcet-Houben M, Hiller E, Rupp S, Gabaldón T, Kovarik P, and Schüller C (2011). Regulation of Candida glabrata oxidative stress resistance is adapted to host environment. FEBS Lett 585(2): 319–327. doi: 10.1016/j.febslet.2010.12.006
- Lelandais G, Tanty V, Geneix C, Etchebest C, Jacq C, and Devaux F (2008). Genome adaptation to chemical stress: clues from comparative transcriptomics in Saccharomyces cerevisiae and Candida glabrata. Genome Biol 9(11): R164. doi: 10.1186/gb-2008-9-11-r164
- Kuo D, Licon K, Bandyopadhyay S, Chuang R, Luo C, Catalana J, Ravasi T, Tan K, and Ideker T (2010). Coevolution within a transcriptional network by compensatory trans and cis mutations. Genome Res 20(12): 1672–1678. doi: 10.1101/gr.111765.110
- Chen K-H, Miyazaki T, Tsai H-F, and Bennett JE (2007). The bZip transcription factor Cgap1p is involved in multidrug resistance and required for activation of multidrug transporter gene CgFLR1 in Candida glabrata. Gene 386(1–2): 63–72. doi: 10.1016/J.GENE.2006.08.010
- Huang M, and Kao KC (2018). Identifying novel genetic determinants for oxidative stress tolerance in Candida glabrata via adaptive laboratory evolution. Yeast 35(11): 605–618. doi: 10.1002/yea.3352
- Brown JA, Sherlock G, Myers CL, Burrows NM, Deng C, Wu HI, McCann KE, Troyanskaya OG, and Brown JM (2006). Global analysis of gene function in yeast by quantitative phenotypic profiling. Mol Syst Biol 2: 2006.0001. doi: 10.1038/msb4100043
- Puig S, Askeland E, and Thiele DJ (2005). Coordinated Remodeling of Cellular Metabolism during Iron Deficiency through Targeted mRNA Degradation. Cell 120(1): 99–110. doi: 10.1016/j.cell.2004.11.032
- Gerwien F, Safyan A, Wisgott S, Hille F, Kaemmer P, Linde J, Brunke S, Kasper L, and Hube B (2016). A novel hybrid iron regulation network combines features from pathogenic and nonpathogenic yeasts. MBio 7(5): e01782-16. doi: 10.1128/mBio.01782-16
- Matsuo R, Mizobuchi S, Nakashima M, Miki K, Ayusawa D, and Fujii M (2017). Central roles of iron in the regulation of oxidative stress in the yeast Saccharomyces cerevisiae. Curr Genet 63(5): 895–907. doi: 10.1007/s00294-017-0689-4
- Csank C, and Haynes K (2000). Candida glabrata displays pseudohyphal growth. FEMS Microbiol Lett 189(1): 115-20. doi: 10.1016/S0378-1097(00)00241-X
- Biswas S, Van Dijck P, and Datta A (2007). Environmental sensing and signal transduction pathways regulating morphopathogenic determinants of Candida albicans. Microbiol Mol Biol Rev 71(2): 348–76. doi: 10.1111/j.1574-6968.2000.tb09216.x
- Brunke S, Seider K, Fischer D, Jacobsen ID, Kasper L, Jablonowski N, Wartenberg A, Bader O, Enache-Angoulvant A, Schaller M, d’Enfert C, and Hube B (2014). One small step for a yeast–microevolution within macrophages renders Candida glabrata hypervirulent due to a single point mutation. PLoS Pathog 10(10): e1004478. doi: 10.1371/journal.ppat.1004478
- Wartenberg A, Linde J, Martin R, Schreiner M, Horn F, Jacobsen ID, Jenull S, Wolf T, Kuchler K, Guthke R, Kurzai O, Forche A, d’Enfert C, Brunke S, and Hube B (2014). Microevolution of Candida albicans in Macrophages Restores Filamentation in a Nonfilamentous Mutant. PLoS Genet 10(12): e1004824. doi: 10.1371/journal.pgen.1004824
- Sudbery PE (2011). Growth of Candida albicans hyphae. Nat Rev Microbiol 9(10): 737–48. doi: 10.1038/nrmicro2636
- Jacobsen ID, Wilson D, Wächtler B, Brunke S, Naglik JR, and Hube B (2012). Candida albicans dimorphism as a therapeutic target. Expert Rev Anti Infect Ther 10(1): 85–93. doi: 10.1586/eri.11.152
- Lo HJ, Köhler JR, DiDomenico B, Loebenberg D, Cacciapuoti A, and Fink GR (1997). Nonfilamentous C. albicans mutants are avirulent. Cell 90(5): 939–49. doi: 10.1016/s0092-8674(00)80358-x
- Doedt T, Krishnamurthy S, Bockmühl DP, Tebarth B, Stempel C, Russell CL, Brown AJP, and Ernst JF (2004). APSES Proteins Regulate Morphogenesis and Metabolism in Candida albicans. Mol Biol Cell 15(7): 3167–3180. doi: 10.1091/mbc.e03-11-0782
- Henry KW, Nickels JT, and Edlind TD (2000). Upregulation of ERG genes in Candida species by azoles and other sterol biosynthesis inhibitors. Antimicrob Agents Chemother 44(10): 2693–700. doi: 10.1128/aac.44.10.2693-2700.2000
- Li QQ, Tsai H-F, Mandal A, Walker BA, Noble JA, Fukuda Y, and Bennett JE (2018). Sterol uptake and sterol biosynthesis act coordinately to mediate antifungal resistance in Candida glabrata under azole and hypoxic stress. Mol Med Rep 17(5): 6585–6597. doi: 10.3892/mmr.2018.8716
- Rogers PD, Vermitsky J-P, Edlind TD, and Hilliard GM (2006). Proteomic analysis of experimentally induced azole resistance in Candida glabrata. J Antimicrob Chemother 58(2): 434–438. doi: 10.1093/jac/dkl221
- dos Santos Silva DB, Carbonera Rodrigues LM, De Almeida AA, de Oliveira KMP, and Grisolia AB (2016). Novel point mutations in the ERG11 gene in clinical isolates of azole resistant Candida species. Mem Inst Oswaldo Cruz 111(3): 192-9. doi: 10.1590/0074-02760150400
- Berila N, Borecka S, Dzugasova V, Bojnansky J, and Subik J (2009). Mutations in the CgPDR1 and CgERG11 genes in azole-resistant Candida glabrata clinical isolates from Slovakia. Int J Antimicrob Agents 33(6): 574–578. doi: 10.1016/j.ijantimicag.2008.11.011
- Shields RK, Kline EG, Healey KR, Kordalewska M, Perlin DS, Nguyen MH, and Clancy CJ (2018). Spontaneous Mutational Frequency and FKS Mutation Rates Vary by Echinocandin Agent against Candida glabrata. Antimicrob Agents Chemother 63(1): e01692-18. doi: 10.1128/AAC.01692-18
ACKNOWLEDGMENTS
Work conducted in this field has been financially supported by “Fundação para a Ciência e a Tecnologia” (FCT) (Contracts PTDC/BBB-BIO/4004/2014 and PTDC/BII-BIO/28216/2017 and PhD scholarships to PP, MG and MC, associated to the BIO-TECNico and AEM FCT PhD programs). Funding received from FCT (grant UID/BIO/04565/2013) and Programa Operacional Regional de Lisboa 2020 (project no. 007317) is also acknowledged.
COPYRIGHT
© 2019

Microevolution of the pathogenic yeasts Candida albicans and Candida glabrata during antifungal therapy and host infection by Pais et al. is licensed under a Creative Commons Attribution 4.0 International License.








