
Viewpoint:
Microbial Cell, Vol. 7, No. 9, pp. 222 - 233; doi: 10.15698/mic2020.09.728
Regulation of the mitochondrial permeability transition pore and its effects on aging

1 AI Lab One, Wilhelmina van Pruisenweg 35, 2595 AN The Hague, The Netherlands.
Keywords: aging, mitochondria, yeast, mPTP, cell death, adenine nucleotide translocator.
Received originally: 31/03/2020 Received in revised form: 12/06/2020
Accepted: 15/06/2020
Published: 22/06/2020
Correspondence:
Damiano Pellegrino-Coppola, AI Lab One, Wilhelmina van Pruisenweg 35, 2595 AN The Hague, The Netherlands; damiano@ailab.one
Conflict of interest statement: DPC is affiliated with AI Lab One.
Please cite this article as: Damiano Pellegrino Coppola (2020). Regulation of the mitochondrial permeability transition pore and its effects on aging. Microbial Cell 7(9): 222-233. doi: 10.15698/mic2020.09.728
Abstract
Aging is an evolutionarily conserved process and is tightly connected to mitochondria. To uncover the aging molecular mechanisms related to mitochondria, different organisms have been extensively used as model systems. Among these, the budding yeast Saccharomyces cerevisiae has been reported multiple times as a model of choice when studying cellular aging. In particular, yeast provides a quick and trustworthy system to identify shared aging genes and pathway patterns. In this viewpoint on aging and mitochondria, I will focus on the mitochondrial permeability transition pore (mPTP), which has been reported and proposed as a main player in cellular aging. I will make several parallelisms with yeast to highlight how this unicellular organism can be used as a guidance system to understand conserved cellular and molecular events in multicellular organisms such as humans. Overall, a thread connecting the preservation of mitochondrial functionality with the activity of the mPTP emerges in the regulation of cell survival and cell death, which in turn could potentially affect aging and aging-related diseases.
INTRODUCTION
Aging can be described as an alteration of the organism in time, characterized by genetical and biochemical dysfunctional changes that ultimately lead to a physical and mental decline and an increased morbidity. The aging process has been clearly linked to many chronological diseases, representing a great risk factor for the development of cardiovascular diseases, neurodegenerations and cancer [1]. Considering that the human population and the mean life expectancy are growing, there is an increasing urge to identify the molecular mechanisms underlying such process, in order to understand the overlap with diseases [2]. However, today the aging core mechanisms still remain poorly understood, and several theories have been proposed to explain the onset and the progression of aging [3]. Indeed, aging is acknowledged as a complex phenomenon encompassing different molecular and cellular players, described as hallmarks of aging [4]. Moreover, these hallmarks can be connected among them, and for this reason aging can be viewed as a cross-pathway process, highlighting that there could be many concurring causes contributing to aging depending on the individual’s background and environmental exposures, and that multiple theories of aging could be right at once, each describing effects of this multifaceted phenomenon from a different perspective. Since genes and pathways associated with these hallmarks have been found as shared with other organisms, aging is also acknowledged as an evolutionary conserved process [5]. Due to the complexity of the aging processes and the average necessary length of the experiments, the budding yeast Saccharomyces cerevisiae has been extensively used as model to understand aging at a cellular level [6]. One of the advantages of using yeast is that aging can be dissected in two features, namely chronological and replicative, that can be used as a model for the aging of post-mitotic and proliferative cells, respectively. In particular, chronological aging is related to quiescent yeast cells in stationary phase and their capacity to remain functional over time (chronological lifespan, CLS), while replicative aging is related to the replicative potential of cells as the number of daughter cells a mother cell can generate (replicative lifespan, RLS; reviewed in [6]). Yeast, which has already proven to be an insightful cellular model in many previous studies, such as for cell cycle and for autophagy [7], also proved its value in aging studies, regarding the anti-aging effects of calorie or diet restriction [8], the inhibition of the Tor pathway [9][10], and by contributing to the discovery of the anti-aging effect of drugs such as spermidine and rapamycin [9][11]. For these reasons, yeast has been proposed as a screening platform to discover anti-aging compounds [12]. Further, yeast has also been extensively employed as a model to study the relation between oxidative stress and aging [13]. Noteworthy, a main anti-aging thread composed of protective mechanisms that deal with reactive oxygen species (ROS) and oxidative stress can be identified. From this perspective, aging can be connected to the mitochondrial free radical theory of aging (MFRTA; [14]) and how ROS, known mainly as byproducts of respiration, can promote mitochondrial dysfunctions and in turn foster and negatively influence the process of aging. Moreover, it is possible that mitochondria indeed play a key role in aging not just through oxidative stress production and protection, but also with other mechanisms and complexes. One complex that has been increasingly acquiring popularity is the mitochondrial permeability transition pore (mPTP), and it has been extensively connected to aging and aging-related diseases (reviewed in [15][16]).
–
In this viewpoint, I will focus on the role of mitochondrial dysfunctions and the mPTP in the context of aging and aging-related diseases, highlighting how evolutionarily conserved molecular mechanisms found in yeast may be used as a guide to understand the molecular mechanisms in humans, and reasoning on the evidence that supports and suggests the mPTP as one of the key regulators of life and death in human cells.
MITOCHONDRIAL DYSFUNCTIONS AND AGING
The MFRTA is a theory derived from the free radical theory of aging [14][17]. In this latter, earlier theory, free radicals are pointed out as the executors of oxidative damage addressed to DNA, proteins and lipids, which in turn causes aging and aging-related diseases [17]. Successively, the theory was extended to mitochondria [14][18], well-known eukaryotic organelles involved in a plethora of cellular events, ranging from metabolism to ATP production, to cell death and more (reviewed in [19][20]). In particular, it is relevant to highlight that mitochondria are responsible for the oxygen-depended ATP production, well-known as oxidative phosphorylation. During this biochemical process, electron leakage occurs, contributing to the creation of ROS through several mitochondrial sites [21]. Although ROS can act as signaling molecules in the modulation of mitochondrial metabolism and fission/fusion mechanisms (reviewed in [22]), an uncontrolled production is acknowledged to be dangerous, and indeed oxidative stress is a feature associated with many diseases, including cardiovascular diseases, cancer and neurodegenerations [23][24]. Initially, mitochondria were thought to be highly vulnerable to ROS, with the mitochondrial DNA (mtDNA) as one main target. Mutations in the mtDNA are also a feature of aging [25], and accumulate in different tissues belonging to the heart [26][27], the skeletal muscles [27], the brain [28][29] and induce senescence [30]. However, mtDNA mutations are not proven to be drivers of aging or aging-related diseases, as it has been shown that DNA polymerase γ-associated proofreading, mtDNA repair systems and mitophagy are efficient systems that prevent an accumulation of damage to the extent required for aging or pathologies [31][32] and mutations in the mtDNA need to accumulate to a certain threshold in order to manifest as a disease [33]. This increase in mutation frequency may be caused by enhanced oxidative stress, but experiments on the antioxidant systems have given controversial results (reviewed in [32]), with the paradox of the naked mole rat which has extensive oxidative stress, but also a remarkably long lifespan [34]. However, homozygous polgmut/mut mice, in which the mitochondrial-specific DNA polymerase γ is mutated and the proofreading activity inactivated, have an accelerated aging phenotype [35]. Further, a DNA damage response-dependent activation of the mTORC1-PGC-1β axis regulates the mitochondrial content contributing to a senescent phenotype [36]. Indeed, the retention of stemness properties has been shown to be the result of an asymmetric division of young mitochondria [37]. Lastly, mitochondrial respiration defects during aging, found to be controlled by epigenetic regulations and not by mtDNA mutations, could be reversed [38]. These results still strongly support a key role of mitochondria in aging, but highlight a different perspective: an increase in ROS and mtDNA mutations, not excluded to be a cause in specific cases, can be generally thought as mechanistically correlated to aging. Overall, this points out that: 1) mitochondria appear as very resilient organelles and 2) the absence of functional mitochondrial activity strongly affects the aging rate.
–
In yeast, an increase in ROS and oxidative stress has also been correlated to aging and lifespan [39], and there is an increase in mtDNA mutations over time [40]. Noteworthy, yeast during proliferation undergoes an asymmetric division that differentiates the daughter cell from the mother cell, with the former inheriting undamaged cellular components, while the latter retains compromised mitochondria and oxidatively damaged proteins [41][42]. Further, special yeast mutants called petite or rho0 lack a functional mitochondrial genome and respiration, and as a result they have a decreased CLS and RLS [43][44]. However, rho0 can undergo metabolic reprogramming, subtelomeric silencing and a mitochondrial adaptation which in turn appears to ameliorate their state and increases their RLS, suggesting that mammalian rho0 can also find a way to potentially adapt and pointing out which mechanisms could be studied [30][45][46][47][48]. Indeed, yeast also highlights that the relation between oxidative stress and cellular lifespan may not be so straightforward as initially thought. In fact, the overexpression of the genes coding for the copper-zinc superoxide dismutase and the manganese superoxide dismutase (SOD1 and SOD2, respectively) increases CLS [49]. On the other hand, inactivation of SOD1 or SOD2 was found to reduce CLS [50][51]. Further, inactivation of SOD1 also dramatically shortens RLS, while inactivation of SOD2 has no remarkable effect [52]. Regarding catalases, the overexpression of CTT1 decreases CLS [49], while its deletion has been shown to increase CLS [53]. The notion that the relation between ROS and aging is not so direct is further suggested by hormesis. Yeast cells exposed to sublethal ROS levels during growth have an extended CLS [54]. This supports the role of ROS as signaling molecules that can act as beneficial, a concept being also explored in mammals, including humans, with ROS involved in adaptive, physiological responses to control metabolism and aging [55][56]. Therefore, the relation between ROS and aging has still to be fully comprehended, in light of the fact that ROS may act as a double-edged sword, therefore it may be that it’s not about ROS per se but it’s related to the level and location of ROS to which cells are exposed [57][58][59].
MITOCHONDRIAL PERMEABILITY TRANSITION PORE AND AGING
A plethora of evidence points to the fact that the proper functioning of mitochondria affects the regulation of lifespan. However, neither mtDNA nor ROS have been uncovered as the ultimate causes of aging. Keeping this in mind, it seems plausible to think about additional players, such as the mPTP, that can potentially fit both health and disease-related mechanisms. The mPTP is a non-selective channel situated in the mitochondrial membrane and which, as the name suggests, is responsible for an increase in the permeability of mitochondria allowing the passage of molecules with a mass up to 1500 Da [60]. Due to its complex multimeric nature, the exact composition and structure of the mPTP is currently under investigation (reviewed in [61][62][63][64]). It is worth mentioning several components: cyclophilin D (CYPD), regarded as a main controller of the opening of the pore; adenine nucleotide translocator (ANT), which is involved in ADP/ATP exchange and in the functioning of the pore; voltage-dependent anion channel (VDAC), located in the outer membrane; ATP synthase, whose role is being intensively discussed [65][66][67]. Over the years, the mPTP opening has been linked to different events, such as Ca2+ overload and oxidative stress [61][62][63][64]. The opening of the pore can hinder mitochondrial metabolism, respiration, and can cause detrimental alterations of the mitochondrial membrane, followed by the release of numerous factors into the cytosol among which Ca2+, NAD+, ROS, AIF, endonuclease G and cytochrome c, causing a fatal variation in cellular homeostasis which has been linked also to aging (reviewed in [15][16][68]). Indeed Ca2+, oxidative stress, decrease in ATP levels and membrane depolarization have all been linked to cellular senescence [69][70][71][72][73]. Loss of NAD+ is also regarded as a feature of aging and further, the disappearance of NAD+ from the cell, subsequent from its release from the mitochondria, can be imputed for instance to the activity of PARP1, involved in the response to DNA damage, or CD38, which has been linked to aging and immune response [15][74][75][76][77]. Lastly, PARP1 hyperactivation, AIF and cytochrome c, are all well known for their role in cell death [78][79][80].
–
A mitochondrial permeability transition resembling peculiar aspects of the mPTP has been observed also in yeast, confirming that the molecular mechanisms of aging can be evolutionarily conserved. In yeast, mitochondria can accumulate Ca2+, increase ROS generation, release cytochrome c and although the identification of the complete structure of the channel responsible for this permeability is still ongoing, multiple yeast orthologs of the mammalian mPTP have been identified [81][82][83][84][85][86][87][88]. This offers the opportunity to understand the functioning of the pore through another cellular system already acknowledged as a model. In particular, the yeast Nuc1 is a mitochondrial mediator involved in apoptosis, ortholog of the mammalian endonuclease G, and its overexpression promotes cells death in presence of H2O2[89]. However, the deletion of the AAC1/2/3 paralog genes for the adenine nucleotide translocators is able to counteract the Nuc1-mediated cell death. A decrease in survival can be observed, but it is possible that in light of their double role as members of the pore and adenine nucleotide translocators, the deletion of the AAC genes impacts cells energetics. Moreover, the isoform of the human adenine nucleotide translocator ANT1 has been shown to confer sensitivity to the pore in presence of H2O2[90]. In fact, ANT1 has been shown to increase the sensitivity of the mPTP, but not to be essential, highlighting that other components may play an important role in the opening of the pore [91]. However, the ANT1 gene is expressed in postmitotic tissues and can be found in the heart and the skeletal muscles, but to a lesser extent also in other organs [92]. Levels are confirmed in the Human Protein Atlas, as ANT1 can be also expressed in parts of the brain and of other organs [93]. Moreover, ANT1 has been linked to different diseases, such as progressive external ophthalmoplegia and mitochondrial DNA depletion syndromes (on Online Mendelian Inheritance in Man: 103220; [94]). Over the years, a link between ANT1, cardiomyopathy and neurodegenerations has been highlighted [15][63][95][96][97][98]. Ultimately, a connection between the mPTP, and in particular ANT1, has been established with cell death and in relation to BAX and BCL2 [99][100][101][102][103][104]. A historical overview on the studies regarding ANT proteins, as well as a collection of structural and functional details over the years has been extensively reviewed [104][105][106].
–
Several parallelisms between the human ANT1 and the yeast ortholog AAC2 can be made: mutations in AAC2, equivalent to the human autosomal dominant progressive external ophthalmoplegia-associated ANT1 mutations, also led yeast to mitochondrial dysfunctions and affected mtDNA [107]. Further, overexpression of the sole ANT1 in yeast did not induce cell death [108], as yeast doesn’t possess ortholog members of the BCL2 family, but yeast mitochondria are still sensible to BAX-mediated apoptosis [109][110], and ANT-deficient yeast cells were shown to be resistant to BAX- [99] and HIV-1 viral protein R-induced apoptosis [111]. Indeed, the yeast Aac proteins have been shown to be required for mitochondria permeabilization and cytochrome c release [112][113].
–
This preliminary evidence strongly suggests that the ANT1-driven cell death runs not only through the mPTP activity, but can be influenced also by pro-apoptotic factors. Always keeping in mind the differences that exist between a model of a system and the system itself, an evolutionarily conserved pattern between yeast and human can be identified, involving adenine nucleotide translocators, mPTP and cell death. Altogether, this highlights a mechanism that appears to be maintained in the history of eukaryotes, indicating that it could represent a main route in the regulation of cell survival and cell death.
MITOCHONDRIAL PERMEABILITY TRANSITION PORE AND ANT1 IN HEALTH AND DISEASE MECHANISMS
Recently, an intriguing connection has been made between the mPTP and autophagy: in particular, in both nematodes and mammals a negative influence of mitochondrial permeability on autophagy-induced lifespan expansion was shown [114]. Indeed mitophagy, the specific degradation of mitochondria, is related to mitochondrial membrane potential, with dysfunctional mitochondria – e.g. depolarized as a consequence of damage – being addressed to mitophagy [115]. Mitophagy of mitochondria has also been recently connected to ANT1, independently from its translocase activity [116]. It is possible that in an autophagy-prone mutant, such as the worm sgk-1, an over-active mPTP causes damages through components involved in mitophagy, and this could explain why the deletion of mitophagy or fragmentation-related genes, such as the fission gene drp-1, had a positive effect on the restoration of lifespan towards normal trends [114].
–
A lowering of the mitochondrial membrane potential is also associated with calorie restriction (CR; reviewed in [117]), and positive effects of CR have been associated with the inhibition of the mPTP (reviewed in [15]). Further, CR is also known to induce autophagy [118] and has been connected to the promotion of mitochondrial fusion [119][120][121], proposed as a countermeasure against excessive mitochondrial degradation during autophagy itself [122]. However, it was also found that mitochondrial fission proteins are increased during CR, and an explanation could be related to the role of fission in mitochondrial biogenesis and CR [123]. Depolarized mitochondria were also found in senescent cells [124][125][126], where autophagy is known to take place [127][128]. In fact, autophagy was found to be activated in senescence although the details of the interplay between autophagy, senescence and apoptosis have to be clarified [128][129]. From this scenario, it is possible to start separating a functional mitochondrial depolarization from hyperpolarization as parts of distinct mechanisms. The first associated with CR, autophagy and senescence, while the latter is associated with an increase in ROS production and apoptosis [130][131], when cells are in a non-cancerous state [132][133][134][135]. In agreement, the role of mitochondrial hyperpolarization in apoptosis was also shown in yeast expressing BAX [136]. Further, the link between hyperpolarization, ROS and cell death in yeast has been proven in many different contexts, such as exposure to H2O2 and cadmium [137][138], or including thermal stress, as reported and summarized in [139], underlying once again the shared biological patterns between this model system and humans.
–
In addition, it has been shown that senescent cells have an increased level of ANT1 [140], and as seen previously, it is also known that an overexpression of ANT1 fosters cell death. However, it is also true that senescent cells are resistant to apoptosis and involved in wound healing [140][141]. Remarkably, the ANT genes have a unique interplay with cell death, as the non-pro-apoptotic gene ANT2 is found in highly proliferative cells such as cancer cells [142][143], while the overexpression of ANT1 induces apoptosis in cancer cells [144]. Of note, TGF-β was shown to repress ANT2 and this contributes to senescence [145][146][147]. On the other hand, in mice the deletion of ANT1 has been shown to bring to hypertrophic cardiomyopathy, while inactivating ANT2 is lethal [148][149][150]. Indeed, fine-tuning of ANT genes and mPTP activity can be both integrated as parts of an interconnected regulatory network involving major cellular pathways in aging, inflammation and diseases, including NF-κB and p53 [142][151][152]. In relation to this, NF-κB was shown to downregulate ANT1 and the activity of mPTP [152][153]. In a hypothetic scenario, NF-κB could therefore promote senescent cell survival, by lowering mitochondrial potential in a functional and ANT1-dependent manner, allowing to send eventual inflammatory and repairing signals, as platelets where found to be activated by senescent cells [154][155]. However, this situation revolves around a delicate equilibrium, as excessive stress and damage could cause an increase in the levels of inflammation, further downregulating ANT1. For instance, this could avoid the leak of ROS from the mPTP, decreasing the level of ROS needed to play a role in the inhibition of proliferation [59][156][157][158], generating an imbalance and fostering a situation where senescent cells drive the origin of cancer cells [159][160][161]. On the other hand, a forced upregulation of ANT1 could cause the opposite situation, where cells are prone to cell death, such as in neurodegenerations where the mPTP has been shown to be involved. In this case, NF-κB could promote the survival of neurons, but glial cells foster inflammation and damage [162][163][164]. Indeed, specific genetic variants could be involved in the modulation of the anti- or pro-death mechanisms. Therefore, ANT1 is a functional and regulatory piece of the mPTP, that is operating as a tap and may influence survival or the execution of a specific type of cell death (Figure 1).
–
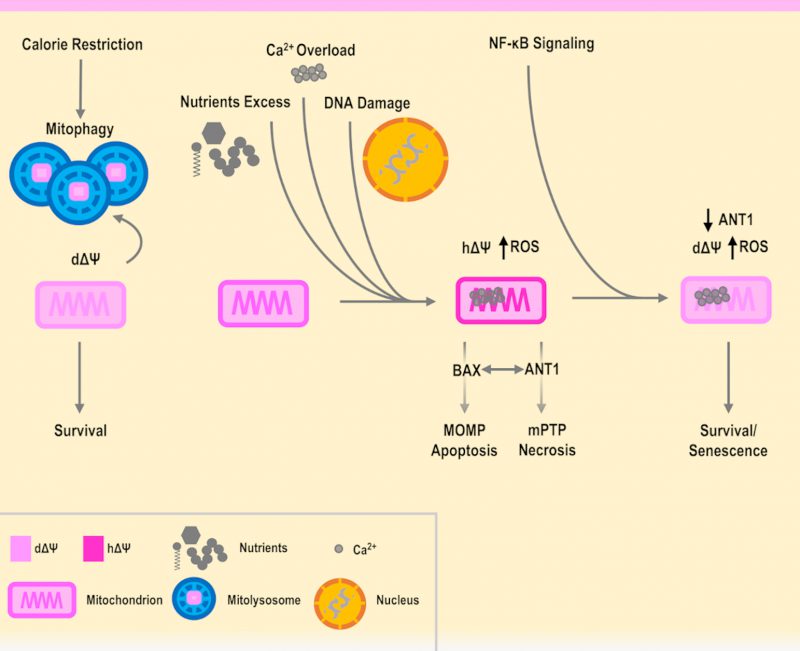 | FIGURE 1: The mPTP, a potential hub in the regulation of cell survival and cell death. Calorie restriction features mitochondrial depolarization and mitophagy, promoting survival. During mitophagy, mitochondria are degraded inside mitolysosomes. In contrast, in presence of death stimuli such as excessive nutrients, calcium overload or DNA damage, cells may undergo cell death. The balance between apoptosis and necrosis could be regulated by the mitochondrial membrane potential coupled with the levels of ANT1 activity and the interactions with other players such as BAX. Further, pathways increasing the expression of ANT1 could foster cell death through the mPTP, while pathways decreasing it (e.g. NF-κB signaling pathway) could favor mPTP closing and cell survival. See the text for details. Δψ, mitochondrial membrane potential with d, depolarized and h, hyperpolarized; MOMP, mitochondrial outer membrane permeabilization; mPTP, mitochondrial permeability transition pore. Organelles, damaged DNA, Ca2+ and nutrients (e.g. carbohydrates such as glucose, proteins, fatty acids) are simplified graphical representations. |
CONCLUSIONS AND FUTURE PERSPECTIVES
The mPTP is regarded as a multimeric complex, of which the complete and definitive structure is currently under investigation. The mPTP has been studied in detail for many years and connected to aging and aging-related diseases, such as neurodegenerations, cardiovascular diseases and cancer. ANT1 appears as a promising candidate to explain the activity of the mPTP, however, it cannot be regarded as exclusive as many other components could have a key role, such as VDAC1 [104][165][166]. An additional player involved in the regulation of mPTP is hexokinase II [167][168][169][170], which has been placed in the context of functional mitochondrial depolarization, described as a key feature of long-lived animals [171]. Both VDAC1 and hexokinase II have orthologs in yeast (Por1 and Hxk2, respectively), and both have been shown to be involved in cell death [112][113][137][172], in agreement with the fact that the budding yeast S. cerevisiae can provide useful information and guidance in the interpretation of the human aging cellular mechanisms. Aging is an evolutionarily conserved process, and shared cellular patterns and pathways are found in eukaryotes ranging from unicellular to multicellular organisms. Evidence accumulated so far can support a beneficial role of functional mitochondria depolarization, as opposed to detrimental hyperpolarization, ROS production and cell death, with the mPTP placed as a regulator in the balance between apoptosis and necrosis (Figure 1). Therefore, based on these premises and employing cellular model systems, on one hand it is necessary to proceed with the identification of the parts that compose the structure of the mPTP and which pathways are involved in its regulation. On the other hand, it would be pivotal to employ different omics technologies and computational methods to trace the expression and the activity of the various mPTP components during aging as well as in health and disease, also in relation to metabolism, epigenetics and the microbiome. For instance, starting from RNA-seq data of whole blood samples from over 3000 individuals of different ages, a recent analysis found ANT1 to be upregulated with aging and nominally significant [173]. Further detailed studies should be planned to consistently confirm this point. Therefore, it is plausible that a threshold could be associated with the mPTP components and activity levels, necessary for a certain aging-related disease to develop, in concert with other molecular cues that govern lifespan and healthspan at the interface between genetic background and environmental exposures.
REFERENCES
- McHugh D, and Gil J (2018). Senescence and aging: Causes, consequences, and therapeutic avenues. J Cell Biol 217(1): 65–77. 10.1083/jcb.201708092
- Hansen M, and Kennedy BK (2016). Does Longer Lifespan Mean Longer Healthspan? Trends Cell Biol 26(8): 565–568. 10.1016/j.tcb.2016.05.002
- Jin K (2010). Modern biological theories of aging. Aging Dis 1(2): 72–74. 10.1093/jn/119.6.952
- López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153(6): 1194. 10.1016/j.cell.2013.05.039
- Bitto A, Wang AM, Bennett CF, and Kaeberlein M (2015). Biochemical Genetic Pathways that Modulate Aging in Multiple Species. Cold Spring Harb Perspect Med 5(11): a025114. 10.1101/cshperspect.a025114
- Longo VD, Shadel GS, Kaeberlein M, and Kennedy B (2012). Replicative and chronological aging in Saccharomyces cerevisiae. Cell Metab 16(1): 18–31. 10.1016/j.cmet.2012.06.002
- Hohmann S (2016). Nobel Yeast Research. FEMS Yeast Res. 16(8): fow094. 10.1093/femsyr/fow094
- Fontana L, Partridge L, and Longo VD (2010). Extending healthy life span-from yeast to humans. Science 328(5976): 321–326. 10.1126/science.1172539
- Powers RW, Kaeberlein M, Caldwell SD, Kennedy BK, and Fields S (2006). Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev 20(2): 174–184. 10.1101/gad.1381406
- Kaeberlein M, Powers RW, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, and Kennedy BK (2005). Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310(5751): 1193–1196. 10.1126/science.1115535
- Eisenberg T, Knauer H, Schauer A, Büttner S, Ruckenstuhl C, Carmona-Gutierrez D, Ring J, Schroeder S, Magnes C, Antonacci L, Fussi H, Deszcz L, Hartl R, Schraml E, Criollo A, Megalou E, Weiskopf D, Laun P, Heeren G, Breitenbach M, Grubeck-Loebenstein B, Herker E, Fahrenkrog B, Fröhlich KU, Sinner F, Tavernarakis N, Minois N, Kroemer G, and Madeo F (2009). Induction of autophagy by spermidine promotes longevity. Nat Cell Biol 11(11): 1305–1314. 10.1038/ncb1975
- Zimmermann A, Hofer S, Pendl T, Kainz K, Madeo F, and Carmona-Gutierrez D (2018). Yeast as a tool to identify anti-aging compounds. FEMS Yeast Res 18(6):foy020. 10.1093/femsyr/foy020
- Dawes IW, and Perrone GG (2020). Stress and ageing in yeast. FEMS Yeast Res 20(1): foz085. 10.1093/femsyr/foz085
- Harman D (1972). The Biologic Clock: The Mitochondria? J Am Geriatr Soc 20(4): 145–147. 10.1111/j.1532-5415.1972.tb00787.x
- Rottenberg H, and Hoek JB (2017). The path from mitochondrial ROS to aging runs through the mitochondrial permeability transition pore. Aging Cell 16(5): 943–955. 10.1111/acel.12650
- Panel M, Ghaleh B, and Morin D (2018). Mitochondria and aging: A role for the mitochondrial transition pore? Aging Cell 17(4): e12793. 10.1111/acel.12793
- Harman D (1956). Aging: A Theory Based on Free Radical and Radiation Chemistry. J Gerontol 11(3): 298–300. 10.1093/geronj/11.3.298
- Harman D (1983). Free radical theory of aging: Consequences of mitochondrial aging. Age 6(3): 86–94. 10.1007/BF02432509
- Eisner V, Picard M, and Hajnóczky G (2018). Mitochondrial dynamics in adaptive and maladaptive cellular stress responses. Nat Cell Biol 20(7): 755–765. 10.1038/s41556-018-0133-0
- Spinelli JB, and Haigis MC (2018). The multifaceted contributions of mitochondria to cellular metabolism. Nat Cell Biol 20(7): 745–754. 10.1038/s41556-018-0124-1
- Wong HS, Dighe PA, Mezera V, Monternier PA, and Brand MD (2017). Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J Biol Chem 292(41): 16804–16809. 10.1074/jbc.R117.789271
- Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, and Griendling KK (2018). Reactive oxygen species in metabolic and inflammatory signaling. Circ Res 122(6): 877–902. 10.1161/CIRCRESAHA.117.311401
- Guillaumet-Adkins A, Yañez Y, Peris-Diaz MD, Calabria I, Palanca-Ballester C, and Sandoval J (2017). Epigenetics and Oxidative Stress in Aging. Oxid Med Cell Longev 2017:9175806. 10.1155/2017/9175806
- Pizzino G, Irrera N, Cucinotta M, Pallio G, Mannino F, Arcoraci V, Squadrito F, Altavilla D, and Bitto A (2017). Oxidative Stress: Harms and Benefits for Human Health. Oxid Med Cell Longev 2017: 8416763. 10.1155/2017/8416763
- Larsson N-G (2010). Somatic Mitochondrial DNA Mutations in Mammalian Aging. Annu Rev Biochem 79(1): 683–706. 10.1146/annurev-biochem-060408-093701
- Corral-Debrinski M, Stepien G, Shoffner JM, Lott MT, Kanter K, and Wallace DC (1991). Hypoxemia Is Associated With Mitochondrial DNA Damage and Gene Induction: Implications for Cardiac Disease. JAMA J Am Med Assoc 266(13): 1812–1816. 10.1001/jama.1991.03470130092035
- Liu VWS, Zhang C, and Nagley P (1998). Mutations in mitochondrial DNA accumulate differentially in three different human tissues during ageing. Nucleic Acids Res 26(5): 1268–1275. 10.1093/nar/26.5.1268
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, and Wallace DC (1992). Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat Genet 2(4): 324–9. 10.1038/ng1292-324
- Soong NW, Hinton DR, Cortopassi G, and Arnheim N (1992). Mosaicism for a specific somatic mitochondrial DNA mutation in adult human brain. Nat Genet 2(4): 318–323. 10.1038/ng1292-318
- Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A, Shirakawa K, Lim HW, Davis SS, Ramanathan A, Gerencser AA, Verdin E, and Campisi J (2016). Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 23(2): 303–14. 10.1016/j.cmet.2015.11.011
- Kauppila TES, Kauppila JHK, and Larsson NG (2017). Mammalian Mitochondria and Aging: An Update. Cell Metab 25(1): 57–71. 10.1016/j.cmet.2016.09.017
- Son JM, and Lee C (2019). Mitochondria: Multifaceted regulators of aging. BMB Rep 52(1): 13–23. 10.5483/BMBRep.2019.52.1.300
- Rossignol R, Faustin B, Rocher C, Malgat M, Mazat JP, and Letellier T (2003). Mitochondrial threshold effects. Biochem J 370(3): 751–762. 10.1042/BJ20021594
- Lewis KN, Andziak B, Yang T, and Buffenstein R (2013). The naked mole-rat response to oxidative stress: just deal with it. Antioxid Redox Signal 19(12): 1388–99. 10.1089/ars.2012.4911
- Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, and Larsson N-G (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429(6990): 417–23. 10.1038/nature02517
- Correia-Melo C, Marques FD, Anderson R, Hewitt G, Hewitt R, Cole J, Carroll BM, Miwa S, Birch J, Merz A, Rushton MD, Charles M, Jurk D, Tait SW, Czapiewski R, Greaves L, Nelson G, Bohlooly-Y M, Rodriguez-Cuenca S, Vidal-Puig A, Mann D, Saretzki G, Quarato G, Green DR, Adams PD, Zglinicki T, Korolchuk VI, and Passos JF (2016). Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35(7): 724–742. 10.15252/embj.201592862
- Katajisto P, Döhla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA, and Sabatini DM (2015). Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science 348(6232): 340–343. 10.1126/science.1260384
- Hashizume O, Ohnishi S, Mito T, Shimizu A, Iashikawa K, Nakada K, Soda M, Mano H, Togayachi S, Miyoshi H, Okita K, and Hayashi JI (2015). Epigenetic regulation of the nuclear-coded GCAT and SHMT2 genes confers human age-associated mitochondrial respiration defects. Sci Rep 5: 10434. 10.1038/srep10434
- Laun P, Pichova A, Madeo F, Fuchs J, Ellinger A, Kohlwein S, Dawes I, Fröhlich KU, and Breitenbach M (2001). Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis. Mol Microbiol 39(5): 1166–1173. 10.1046/j.1365-2958.2001.02317.x
- Veatch JR, McMurray MA, Nelson ZW, and Gottschling DE (2009). Mitochondrial Dysfunction Leads to Nuclear Genome Instability via an Iron-Sulfur Cluster Defect. Cell 137(7): 1247–1258. 10.1016/j.cell.2009.04.014
- Aguilaniu H, Gustafsson L, Rigoulet M, and Nyström T (2003). Asymmetric inheritance of oxidatively damaged proteins during cytokinesis. Science 299(5613): 1751–1753. 10.1126/science.1080418
- Vevea JD, Swayne TC, Boldogh IR, and Pon LA (2014). Inheritance of the fittest mitochondria in yeast. Trends Cell Biol 24(1): 53–60. 10.1016/j.tcb.2013.07.003
- Ocampo A, Liu J, Schroeder EA, Shadel GS, and Barrientos A (2012). Mitochondrial respiratory thresholds regulate yeast chronological life span and its extension by caloric restriction. Cell Metab 16(1): 55–67. 10.1016/j.cmet.2012.05.013
- Yi D-G, Hong S, and Huh W-K (2018). Mitochondrial dysfunction reduces yeast replicative lifespan by elevating RAS-dependent ROS production by the ER-localized NADPH oxidase Yno1. PLoS One 13(6): e0198619. 10.1371/journal.pone.0198619
- Fernández-Moreno M, Hermida-Gómez T, Gallardo ME, Dalmao-Fernández A, Rego-Pérez I, Garesse R, and Blanco FJ (2016). Generating Rho-0 Cells Using Mesenchymal Stem Cell Lines. PLoS One 11(10): e0164199. 10.1371/journal.pone.0164199
- Garcia EJ, de Jonge JJ, Liao P-C, Stivison E, Sing CN, Higuchi-Sanabria R, Boldogh IR, and Pon LA (2019). Reciprocal interactions between mtDNA and lifespan control in budding yeast. Mol Biol Cell 30(24): 2943–2952. 10.1091/mbc.E18-06-0356
- Guaragnella N, Coyne LP, Chen XJ, and Giannattasio S (2018). Mitochondria–cytosol–nucleus crosstalk: learning from Saccharomyces cerevisiae. FEMS Yeast Res 18(8): foy088. 10.1093/FEMSYR/FOY088
- Jazwinski SM, Jiang JC, and Kim S (2018). Adaptation to metabolic dysfunction during aging: Making the best of a bad situation. Exp Gerontol 107: 87–90. 10.1016/j.exger.2017.07.013
- Fabrizio P, Liou LL, Moy VN, Diaspro A, Valentine JS, Gralla EB, and Longo VD (2003). SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics 163(1): 35–46. 12586694
- Feser J, Truong D, Das C, Carson JJ, Kieft J, Harkness T, and Tyler JK (2010). Elevated Histone Expression Promotes Life Span Extension. Mol Cell 39(5): 724–735. 10.1016/j.molcel.2010.08.015
- Weinberger M, Mesquita A, Carroll T, Marks L, Yang H, Zhang Z, Ludovico P, and Burhans WC (2010). Growth signaling promotes chronological aging in budding yeast by inducing superoxide anions that inhibit quiescence. Aging 2(10): 709. https://doi.org/10.18632/aging.100215
- Kaeberlein M, Kirkland KT, Fields S, and Kennedy BK (2005). Genes determining yeast replicative life span in a long-lived genetic background. Mech Ageing Dev 126(4): 491–504. 10.1016/j.mad.2004.10.007
- Mesquita A, Weinberger M, Silva A, Sampaio-Marques B, Almeida B, Leão C, Costa V, Rodrigues F, Burhans WC, and Ludovico P (2010). Caloric restriction or catalase inactivation extends yeast chronological lifespan by inducing H2O2 and superoxide dismutase activity. Proc Natl Acad Sci U S A 107(34): 15123–15128. 10.1073/pnas.1004432107
- Pan Y, Schroeder EA, Ocampo A, Barrientos A, and Shadel GS (2011). Regulation of yeast chronological life span by TORC1 via adaptive mitochondrial ROS signaling. Cell Metab 13(6): 668–78. 10.1016/j.cmet.2011.03.018
- Ristow M (2014). Unraveling the truth about antioxidants: mitohormesis explains ROS-induced health benefits. Nat Med 20(7): 709–11. 10.1038/nm.3624
- Shadel GS, and Horvath TL (2015). Mitochondrial ROS Signaling in Organismal Homeostasis. Cell 163(3): 560–569. 10.1016/j.cell.2015.10.001
- Chandel NS, and Tuveson DA (2014). The promise and perils of antioxidants for cancer patients. N Engl J Med 371(2): 177–8. 10.1056/NEJMcibr1405701
- Mittler R (2017). ROS Are Good. Trends Plant Sci 22(1): 11–19. 10.1016/j.tplants.2016.08.002
- Diebold L, and Chandel NS (2016). Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med 100: 86–93. 10.1016/j.freeradbiomed.2016.04.198
- Haworth RA, and Hunter DR (1979). The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 195(2): 460–467. 10.1016/0003-9861(79)90372-2
- Bauer TM, and Murphy E (2020). Role of Mitochondrial Calcium and the Permeability Transition Pore in Regulating Cell Death. Circ Res 126(2): 280–293. 10.1161/CIRCRESAHA.119.316306
- Karch J, and Molkentin JD (2018). Identity of the elusive mitochondrial permeability transition pore: what it might be, what it was, and what it still could be. Curr Opin Physiol 3: 57–62. 10.1016/j.cophys.2018.03.001
- Halestrap AP (2009). What is the mitochondrial permeability transition pore? J Mol Cell Cardiol 46(6): 821–831. 10.1016/j.yjmcc.2009.02.021
- Biasutto L, Azzolini M, Szabò I, and Zoratti M (2016). The mitochondrial permeability transition pore in AD 2016: An update. Biochim Biophys Acta – Mol Cell Res 1863(10): 2515–2530. 10.1016/j.bbamcr.2016.02.012
- Bernardi P (2018). Why F-ATP Synthase Remains a Strong Candidate as the Mitochondrial Permeability Transition Pore. Front Physiol 9(NOV): 1543. 10.3389/fphys.2018.01543
- Carroll J, He J, Ding S, Fearnley IM, and Walker JE (2019). Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc Natl Acad Sci U S A 116(26): 12816–12821. 10.1073/pnas.1904005116
- Urbani A, Giorgio V, Carrer A, Franchin C, Arrigoni G, Jiko C, Abe K, Maeda S, Shinzawa-Itoh K, Bogers JFM, McMillan DGG, Gerle C, Szabò I, and Bernardi P (2019). Purified F-ATP synthase forms a Ca2+-dependent high-conductance channel matching the mitochondrial permeability transition pore. Nat Commun 10(1): 4341. 10.1038/s41467-019-12331-1
- Kroemer G, Galluzzi L, and Brenner C (2007). Mitochondrial membrane permeabilization in cell death. Physiol Rev 87(1): 99–163. 10.1152/physrev.00013.2006
- Wiel C, Lallet-Daher H, Gitenay D, Gras B, Le Calvé B, Augert A, Ferrand M, Prevarskaya N, Simonnet H, Vindrieux D, and Bernard D (2014). Endoplasmic reticulum calcium release through ITPR2 channels leads to mitochondrial calcium accumulation and senescence. Nat Commun 5: 3792. 10.1038/ncomms4792
- Stöckl P, Zankl C, Hütter E, Unterluggauer H, Laun P, Heeren G, Bogengruber E, Herndler-Brandstetter D, Breitenbach M, and Jansen-Dürr P (2007). Partial uncoupling of oxidative phosphorylation induces premature senescence in human fibroblasts and yeast mother cells. Free Radic Biol Med 43(6): 947–58. 10.1016/j.freeradbiomed.2007.06.005
- Ziegler D V, Wiley CD, and Velarde MC (2015). Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14(1): 1–7. 10.1111/acel.12287
- Dumont P, Burton M, Chen QM, Gonos ES, Frippiat C, Mazarati JB, Eliaers F, Remacle J, and Toussaint O (2000). Induction of replicative senescence biomarkers by sublethal oxidative stresses in normal human fibroblast. Free Radic Biol Med 28(3): 361–373. 10.1016/S0891-5849(99)00249-X
- Kornienko JS, Smirnova IS, Pugovkina NA, Ivanova JS, Shilina MA, Grinchuk TM, Shatrova AN, Aksenov ND, Zenin V V, Nikolsky NN, and Lyublinskaya OG (2019). High doses of synthetic antioxidants induce premature senescence in cultivated mesenchymal stem cells. Sci Rep 9(1): 1296. 10.1038/s41598-018-37972-y
- Camacho-Pereira J, Tarragó MG, Chini CCS, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, and Chini EN (2016). CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab 23(6): 1127–1139. 10.1016/j.cmet.2016.05.006
- Hogan KA, Chini CCS, and Chini EN (2019). The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front Immunol 10(MAY): 1187. 10.3389/fimmu.2019.01187
- Schriewer JM, Peek CB, Bass J, and Schumacker PT (2013). ROS-mediated PARP activity undermines mitochondrial function after permeability transition pore opening during myocardial ischemia-reperfusion. J Am Heart Assoc 2(2): e000159. 10.1161/JAHA.113.000159
- Kahraman S, Siegel A, Polster BM, and Fiskum G (2015). Permeability transition pore-dependent and PARP-mediated depletion of neuronal pyridine nucleotides during anoxia and glucose deprivation. J Bioenerg Biomembr 47(1–2): 53–61. 10.1007/s10863-014-9588-2
- Morales JC, Li L, Fattah FJ, Dong Y, Bey EA, Patel M, Gao J, and Boothman DA (2014). Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit Rev Eukaryot Gene Expr 24(1): 15–28. 10.1615/CritRevEukaryotGeneExpr.2013006875
- Alano CC, Ying W, and Swanson RA (2004). Poly(ADP-ribose) polymerase-1-mediated cell death in astrocytes requires NAD+ depletion and mitochondrial permeability transition. J Biol Chem 279(18): 18895–902. 10.1074/jbc.M313329200
- Cipriani G, Rapizzi E, Vannacci A, Rizzuto R, Moroni F, and Chiarugi A (2005). Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J Biol Chem 280(17): 17227–34. 10.1074/jbc.M414526200
- Azzolin L, von Stockum S, Basso E, Petronilli V, Forte MA, and Bernardi P (2010). The mitochondrial permeability transition from yeast to mammals. FEBS Lett 584(12): 2504–2509. 10.1016/j.febslet.2010.04.023
- Jung DW, Bradshaw PC, and Pfeiffer DR (1997). Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J Biol Chem 272(34): 21104–21112. 10.1074/jbc.272.34.21104
- Manon S, Roucou X, Guérin M, Rigoulet M, and Guérin B (1998). Minireview: Characterization of the yeast mitochondria unselective channel: A counterpart to the mammalian permeability transition pore? J Bioenerg Biomembr 30(5): 419–429. 10.1023/A:1020533928491
- Yamada A, Yamamoto T, Yoshimura Y, Gouda S, Kawashima S, Yamazaki N, Yamashita K, Kataoka M, Nagata T, Terada H, Pfeiffer DR, and Shinohara Y (2009). Ca2+-induced permeability transition can be observed even in yeast mitochondria under optimized experimental conditions. Biochim Biophys Acta – Bioenerg 1787(12): 1486–1491. 10.1016/j.bbabio.2009.07.001
- Vianello A, Casolo V, Petrussa E, Peresson C, Patui S, Bertolini A, Passamonti S, Braidot E, and Zancani M (2012). The mitochondrial permeability transition pore (PTP) – An example of multiple molecular exaptation? Biochim Biophys Acta – Bioenerg 1817(11): 2072–2086. 10.1016/j.bbabio.2012.06.620
- Carraro M, Giorgio V, Sileikyte J, Sartori G, Forte M, Lippe G, Zoratti M, Szabò I, and Bernardi P (2014). Channel formation by yeast F-ATP synthase and the role of dimerization in the mitochondrial permeability transition. J Biol Chem 289(23): 15980–15985. 10.1074/jbc.C114.559633
- Carraro M, and Bernardi P (2016). Calcium and reactive oxygen species in regulation of the mitochondrial permeability transition and of programmed cell death in yeast. Cell Calcium 60(2): 102–107. 10.1016/j.ceca.2016.03.005
- Carraro M, Checchetto V, Sartori G, Kucharczyk R, di Rago J-P, Minervini G, Franchin C, Arrigoni G, Giorgio V, Petronilli V, Tosatto SCE, Lippe G, Szabó I, and Bernardi P (2018). High-Conductance Channel Formation in Yeast Mitochondria is Mediated by F-ATP Synthase e and g Subunits. Cell Physiol Biochem 50(5): 1840–1855. 10.1159/000494864
- Büttner S, Eisenberg T, Carmona-Gutierrez D, Ruli D, Knauer H, Ruckenstuhl C, Sigrist C, Wissing S, Kollroser M, Fröhlich KU, Sigrist S, and Madeo F (2007). Endonuclease G Regulates Budding Yeast Life and Death. Mol Cell 25(2): 233–246. 10.1016/j.molcel.2006.12.021
- Doczi J, Torocsik B, Echaniz-Laguna A, Mousson de Camaret B, Starkov A, Starkova N, Gál A, Molnár MJ, Kawamata H, Manfredi G, Adam-Vizi V, and Chinopoulos C (2016). Alterations in voltage-sensing of the mitochondrial permeability transition pore in ANT1-deficient cells. Sci Rep 6: 26700. 10.1038/srep26700
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, and Wallace DC (2004). The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427(6973): 461–5. 10.1038/nature02229
- Dolce V, Scarcia P, Iacopetta D, and Palmieri F (2005). A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett 579(3): 633–7. 10.1016/j.febslet.2004.12.034
- Uhlén M et al. (2015). Tissue-based map of the human proteome. Science 347(6220). 10.1126/science.1260419
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, and Hamosh A (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic Acids Res 43(Database issue): D789-98. 10.1093/nar/gku1205
- Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, Narula N, Dreike S, Platt J, Procaccio V, Ortiz-González XR, Puffenberger EG, Kelley RI, Morton DH, Narula J, and Wallace DC (2013). Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci U S A 110(9): 3453–8. 10.1073/pnas.1300690110
- Lee J, Schriner SE, and Wallace DC (2009). Adenine nucleotide translocator 1 deficiency increases resistance of mouse brain and neurons to excitotoxic insults. Biochim Biophys Acta 1787(5): 364–70. 10.1016/j.bbabio.2009.01.014
- Dörner A, and Schultheiss HP (2000). The myocardial expression of the adenine nucleotide translocator isoforms is specifically altered in dilated cardiomyopathy. Herz 25(3): 176–180. 10.1007/s000590050004
- Schultheiss HP, Schulze K, and Dörner A (1996). Significance of the adenine nucleotide translocator in the pathogenesis of viral heart disease. Mol Cell Biochem 163–164(1): 319–327. 10.1007/BF00408672
- Marzo I, Brenner C, Zamzami N, Jürgensmeier JM, Susin SA, Vieira HL, Prévost MC, Xie Z, Matsuyama S, Reed JC, and Kroemer G (1998). Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis. Science 281(5385): 2027–31. 10.1126/science.281.5385.2027
- Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, and Kroemer G (1999). Molecular characterization of mitochodrial apoptosis-inducing factor. Nature 397(6718): 441–446. 10.1038/17135
- Belzacq AS, Vieira HLA, Verrier F, Vandecasteele G, Cohen I, Prévost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, and Kroemer G (2003). Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res 63(2): 541–546. 12543814
- Baines CP, and Molkentin JD (2009). Adenine nucleotide translocase-1 induces cardiomyocyte death through upregulation of the pro-apoptotic protein Bax. J Mol Cell Cardiol 46(6): 969–77. 10.1016/j.yjmcc.2009.01.016
- Zhivotovsky B, Galluzzi L, Kepp O, and Kroemer G (2009). Adenine nucleotide translocase: a component of the phylogenetically conserved cell death machinery. Cell Death Differ 16(11): 1419–25. 10.1038/cdd.2009.118
- Grimm S, and Brdiczka D (2007). The permeability transition pore in cell death. Apoptosis 12(5): 841–855. 10.1007/s10495-007-0747-3
- Halestrap AP, and Brenner C (2003). The Adenine Nucleotide Translocase: A Central Component of the Mitochondrial Permeability Transition Pore and Key Player in Cell Death. Curr Med Chem 10: 1507–1525. 10.2174/0929867033457278
- Klingenberg M (2008). The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta – Biomembr 1778(10): 1978–2021. 10.1016/j.bbamem.2008.04.011
- Fontanesi F, Palmieri L, Scarcia P, Lodi T, Donnini C, Limongelli A, Tiranti V, Zeviani M, Ferrero I, and Viola AM (2004). Mutations in AAC2, equivalent to human adPEO-associated ANT1 mutations, lead to defective oxidative phosphorylation in Saccharomyces cerevisiae and affect mitochondrial DNA stability. Hum Mol Genet 13(9): 923–934. 10.1093/hmg/ddh108
- Bauer MK, Schubert A, Rocks O, and Grimm S (1999). Adenine nucleotide translocase-1, a component of the permeability transition pore, can dominantly induce apoptosis. J Cell Biol 147(7): 1493–502. 10.1083/jcb.147.7.1493
- Sato T, Hanada M, Bodrug S, Irie S, Iwama N, Boise LH, Thompson CB, Golemis E, Fong L, Wang HG, and Reed JC (1994). Interactions among members of the Bcl-2 protein family analyzed with a yeast two-hybrid system. Proc Natl Acad Sci U S A 91(20): 9238–9242. 10.1073/pnas.91.20.9238
- Polčic P, Jaká P, and Mentel M (2015). Yeast as a tool for studying proteins of the Bcl-2 family. Microbial Cell 2(3): 74–87. 10.15698/mic2015.03.193
- Jacotot E, Ravagnan L, Loeffler M, Ferri KF, Vieira HLA, Zamzami N, Costantini P, Druillennec S, Hoebeke J, Briand JP, Irinopoulou T, Daugas E, Susin SA, Cointe D, Xie ZH, Reed JC, Roques BP, and Kroemer G (2000). The HIV-1 viral protein R induces apoptosis via a direct effect on the mitochondrial permeability transition pore. J Exp Med 191(1): 33–45. 10.1084/jem.191.1.33
- Pereira C, Camougrand N, Manon S, Sousa MJ, and Côrte-Real M (2007). ADP/ATP carrier is required for mitochondrial outer membrane permeabilization and cytochrome c release in yeast apoptosis. Mol Microbiol 66(3): 571–582. 10.1111/j.1365-2958.2007.05926.x
- Trindade D, Pereira C, Chaves SR, Manon S, Côrte-Real M, and Sousa MJ (2016). VDAC regulates AAC-mediated apoptosis and cytochrome c release in yeast. Microbial Cell 3(10): 500–510. 10.15698/mic2016.10.533
- Zhou B, Kreuzer J, Kumsta C, Wu L, Kamer KJ, Cedillo L, Zhang Y, Li S, Kacergis MC, Webster CM, Fejes-Toth G, Naray-Fejes-Toth A, Das S, Hansen M, Haas W, and Soukas AA (2019). Mitochondrial Permeability Uncouples Elevated Autophagy and Lifespan Extension. Cell 177(2): 299-314.e16. 10.1016/j.cell.2019.02.013
- Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, and Bohr VA (2016). Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 17(5): 308–321. 10.1038/nrm.2016.14
- Hoshino A, Wang W, Wada S, McDermott-Roe C, Evans CS, Gosis B, Morley MP, Rathi KS, Li J, Li K, Yang S, McManus MJ, Bowman C, Potluri P, Levin M, Damrauer S, Wallace DC, Holzbaur ELF, and Arany Z (2019). The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 575(7782): 375–379. 10.1038/s41586-019-1667-4
- Martin-Montalvo A, and De Cabo R (2013). Mitochondrial metabolic reprogramming induced by calorie restriction. Antioxidants Redox Signal 19(3): 310–320. 10.1089/ars.2012.4866
- Madeo F, Carmona-Gutierrez D, Hofer SJ, and Kroemer G (2019). Caloric Restriction Mimetics against Age-Associated Disease: Targets, Mechanisms, and Therapeutic Potential. Cell Metab 29(3): 592–610. 10.1016/J.CMET.2019.01.018
- Greenberg EF, and Vatolin S (2018). Symbiotic Origin of Aging. Rejuvenation Res 21(3): 225–231. 10.1089/rej.2017.1973
- Liesa M, and Shirihai OS (2013). Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab 17(4): 491–506. 10.1016/J.CMET.2013.03.002
- Chaudhari SN, and Kipreos ET (2017). Increased mitochondrial fusion allows the survival of older animals in diverse C. elegans longevity pathways. Nat Commun 8(1): 182. 10.1038/s41467-017-00274-4
- Galluzzi L, Kepp O, and Kroemer G (2011). Mitochondrial Dynamics: A Strategy for Avoiding Autophagy. Curr Biol 21(12): R478–R480. 10.1016/J.CUB.2011.05.002
- Putti R, Sica R, Migliaccio V, and Lionetti L (2015). Diet impact on mitochondrial bioenergetics and dynamics. Front Physiol 6: 109. 10.3389/fphys.2015.00109
- Passos JF, Saretzki G, Ahmed S, Nelson G, Richter T, Peters H, Wappler I, Birket MJ, Harold G, Schaeuble K, Birch-Machin MA, Kirkwood TBL, and von Zglinicki T (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol 5(5): e110. 10.1371/journal.pbio.0050110
- Passos JF, Nelson G, Wang C, Richter T, Simillion C, Proctor CJ, Miwa S, Olijslagers S, Hallinan J, Wipat A, Saretzki G, Rudolph KL, Kirkwood TBL, and Von Zglinicki T (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol Syst Biol 6: 347. 10.1038/msb.2010.5
- Studencka M, and Schaber J (2017). Senoptosis: non-lethal DNA cleavage as a route to deep senescence. Oncotarget 8(19): 30656–30671. 10.18632/oncotarget.15693
- Young ARJ, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JFJ, Tavaré S, Arakawa S, Shimizu S, Watt FM, and Narita M (2009). Autophagy mediates the mitotic senescence transition. Genes Dev 23(7): 798–803. 10.1101/gad.519709
- Kwon Y, Kim JW, Jeoung JA, Kim MS, and Kang C (2017). Autophagy is pro-senescence when seen in close-up, but anti-senescence in long-shot. Mol Cells 40(9): 607–612. 10.14348/molcells.2017.0151
- Doherty J, and Baehrecke EH (2018). Life, death and autophagy. Nat Cell Biol 20(10): 1110–1117. 10.1038/s41556-018-0201-5
- Perl A, Gergely P, Nagy G, Koncz A, and Banki K (2004). Mitochondrial hyperpolarization: A checkpoint of T-cell life, death and autoimmunity. Trends Immunol 25(7): 360–367. 10.1016/j.it.2004.05.001
- Gergely P, Niland B, Gonchoroff N, Pullmann R, Phillips PE, and Perl A (2002). Persistent mitochondrial hyperpolarization, increased reactive oxygen intermediate production, and cytoplasmic alkalinization characterize altered IL-10 signaling in patients with systemic lupus erythematosus. J Immunol 169(2): 1092–101. 10.4049/jimmunol.169.2.1092
- Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, and Michelakis ED (2007). A Mitochondria-K+ Channel Axis Is Suppressed in Cancer and Its Normalization Promotes Apoptosis and Inhibits Cancer Growth. Cancer Cell 11(1): 37–51. 10.1016/j.ccr.2006.10.020
- Heerdt BG, Houston MA, and Augenlicht LH (2005). The intrinsic mitochondrial membrane potential of colonic carcinoma cells is linked to the probability of tumor progression. Cancer Res 65(21): 9861–9867. 10.1158/0008-5472.CAN-05-2444
- Heerdt BG, Houston MA, Wilson AJ, and Augenlicht LH (2003). The intrinsic mitochondrial membrane potential (δψm) is associated with steady-state mitochondrial activity and the extent to which colonic epithelial cells undergo butyrate-mediated growth arrest and apoptosis. Cancer Res 63(19): 6311–6319. 14559818
- Schieke SM, Ma M, Cao L, McCoy JP, Liu C, Hensel NF, Barrett AJ, Boehm M, and Finkel T (2008). Mitochondrial metabolism modulates differentiation and teratoma formation capacity in mouse embryonic stem cells. J Biol Chem 283(42): 28506–28512. 10.1074/jbc.M802763200
- Gross A, Pilcher K, Blachly-Dyson E, Basso E, Jockel J, Bassik MC, Korsmeyer SJ, and Forte M (2000). Biochemical and Genetic Analysis of the Mitochondrial Response of Yeast to BAX and BCL-XL. Mol Cell Biol 20(9): 3125–3136. 10.1128/mcb.20.9.3125-3136.2000
- Amigoni L, Frigerio G, Martegani E, and Colombo S (2016). Involvement of Aif1 in apoptosis triggered by lack of Hxk2 in the yeast Saccharomyces cerevisiae. FEMS Yeast Res 16(3): fow016. 10.1093/femsyr/fow016
- Nargund AM, Avery S V., and Houghton JE (2008). Cadmium induces a heterogeneous and caspase-dependent apoptotic response in Saccharomyces cerevisiae. Apoptosis 13(6): 811–821. 10.1007/s10495-008-0215-8
- Fedoseeva I V., Pyatrikas D V., Stepanov A V., Fedyaeva A V., Varakina NN, Rusaleva TM, Borovskii GB, and Rikhvanov EG (2017). The role of flavin-containing enzymes in mitochondrial membrane hyperpolarization and ROS production in respiring Saccharomyces cerevisiae cells under heat-shock conditions. Sci Rep 7(1). 10.1038/s41598-017-02736-7
- Hubackova S, Davidova E, Rohlenova K, Stursa J, Werner L, Andera L, Dong L, Terp MG, Hodny Z, Ditzel HJ, Rohlena J, and Neuzil J (2019). Selective elimination of senescent cells by mitochondrial targeting is regulated by ANT2. Cell Death Differ26(2): 276–290. 10.1038/s41418-018-0118-3
- Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR, Laberge RM, Vijg J, VanSteeg H, Dollé MET, Hoeijmakers JHJ, deBruin A, Hara E, and Campisi J (2014). An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 31(6): 722–733. 10.1016/j.devcel.2014.11.012
- Zamora M, Meroño C, Viñas O, and Mampel T (2004). Recruitment of NF-kappaB into mitochondria is involved in adenine nucleotide translocase 1 (ANT1)-induced apoptosis. J Biol Chem 279(37): 38415–23. 10.1074/jbc.M404928200
- Chevrollier A, Loiseau D, Reynier P, and Stepien G (2011). Adenine nucleotide translocase 2 is a key mitochondrial protein in cancer metabolism. Biochim Biophys Acta – Bioenerg 1807(6): 562–567. 10.1016/j.bbabio.2010.10.008
- Jang J-Y, Choi Y, Jeon Y-K, Aung KCY, and Kim C-W (2008). Over-expression of adenine nucleotide translocase 1 (ANT1) induces apoptosis and tumor regression in vivo. BMC Cancer 8: 160. 10.1186/1471-2407-8-160
- Luciakova K, Kollarovic G, Kretova M, Šabova L, and Nelson BD (2011). TGF-β signals the formation of a unique NF1/Smad4-dependent transcription repressor-complex in human diploid fibroblasts. Biochem Biophys Res Commun 411(3): 648–653. 10.1016/j.bbrc.2011.07.017
- Kretova M, Sabova L, Hodny Z, Bartek J, Kollarovic G, Nelson BD, Hubackova S, and Luciakova K (2014). TGF-β/NF1/Smad4-mediated suppression of ANT2 contributes to oxidative stress in cellular senescence. Cell Signal 26(12): 2903–2911. 10.1016/j.cellsig.2014.08.029
- Hubackova S, Kucerova A, Michlits G, Kyjacova L, Reinis M, Korolov O, Bartek J, and Hodny Z (2016). IFNγ induces oxidative stress, DNA damage and tumor cell senescence via TGFβ/SMAD signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene 35(10): 1236–1249. 10.1038/onc.2015.162
- Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, and Wallace DC (1997). A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet 16(3): 226–34. 10.1038/ng0797-226
- Narula N, Zaragoza M V, Sengupta PP, Li P, Haider N, Verjans J, Waymire K, Vannan M, and Wallace DC (2011). Adenine nucleotide translocase 1 deficiency results in dilated cardiomyopathy with defects in myocardial mechanics, histopathological alterations, and activation of apoptosis. JACC Cardiovasc Imaging 4(1): 1–10. 10.1016/j.jcmg.2010.06.018
- Kokoszka JE, Waymire KG, Flierl A, Sweeney KM, Angelin A, MacGregor GR, and Wallace DC (2016). Deficiency in the mouse mitochondrial adenine nucleotide translocator isoform 2 gene is associated with cardiac noncompaction. Biochim Biophys Acta 1857(8): 1203–1212. 10.1016/j.bbabio.2016.03.026
- Vaseva A V, Marchenko ND, Ji K, Tsirka SE, Holzmann S, and Moll UM (2012). p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 149(7): 1536–48. 10.1016/j.cell.2012.05.014
- Zhang C, Jiang H, Wang P, Liu H, and Sun X (2017). Transcription factor NF-kappa B represses ANT1 transcription and leads to mitochondrial dysfunctions. Sci Rep 7: 44708. 10.1038/srep44708
- Dhingra R, Guberman M, Rabinovich-Nikitin I, Gerstein J, Margulets V, Gang H, Madden N, Thliveris J, and Kirshenbaum LA (2019). Impaired NF-κB signalling underlies cyclophilin D-mediated mitochondrial permeability transition pore opening in doxorubicin cardiomyopathy. Cardiovasc Res10.1093/cvr/cvz240
- Wiley CD, Liu S, Limbad C, Zawadzka AM, Beck J, Demaria M, Artwood R, Alimirah F, Lopez-Dominguez J-A, Kuehnemann C, Danielson SR, Basisty N, Kasler HG, Oron TR, Desprez P-Y, Mooney SD, Gibson BW, Schilling B, Campisi J, and Kapahi P (2019). SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep 28(13): 3329-3337.e5. 10.1016/j.celrep.2019.08.049
- Valenzuela CA, Quintanilla R, Olate-Briones A, Venturini W, Mancilla D, Cayo A, Moore-Carrasco R, and Brown NE (2019). SASP-Dependent Interactions between Senescent Cells and Platelets Modulate Migration and Invasion of Cancer Cells. Int J Mol Sci 20(21). 10.3390/ijms20215292
- Hamanaka RB, and Chandel NS (2010). Mitochondrial reactive oxygen species regulate cellular signaling and dictate biological outcomes. Trends Biochem Sci 35(9): 505–513. 10.1016/j.tibs.2010.04.002
- Topf U, Suppanz I, Samluk L, Wrobel L, Böser A, Sakowska P, Knapp B, Pietrzyk MK, Chacinska A, and Warscheid B (2018). Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat Commun 9(1): 324. 10.1038/s41467-017-02694-8
- Schieber M, and Chandel NS (2014). ROS function in redox signaling and oxidative stress. Curr Biol 24(10). 10.1016/j.cub.2014.03.034
- Lee S, and Schmitt CA (2019). The dynamic nature of senescence in cancer. Nat Cell Biol 21(1): 94–101. 10.1038/s41556-018-0249-2
- Cruickshanks HA, McBryan T, Nelson DM, Vanderkraats ND, Shah PP, van Tuyn J, Singh Rai T, Brock C, Donahue G, Dunican DS, Drotar ME, Meehan RR, Edwards JR, Berger SL, and Adams PD (2013). Senescent cells harbour features of the cancer epigenome. Nat Cell Biol 15(12): 1495–506. 10.1038/ncb2879
- Faget D V, Ren Q, and Stewart SA (2019). Unmasking senescence: context-dependent effects of SASP in cancer. Nat Rev Cancer 19(8): 439–453. 10.1038/s41568-019-0156-2
- Camandola S, and Mattson MP (2007). NF-κB as a therapeutic target in neurodegenerative diseases. Expert Opin Ther Targets 11(2): 123–132. 10.1517/14728222.11.2.123
- Mattson MP, and Meffert MK (2006). Roles for NF-κB in nerve cell survival, plasticity, and disease. Cell Death Differ 13(5): 852–860. 10.1038/sj.cdd.4401837
- Dresselhaus EC, and Meffert MK (2019). Cellular specificity of NF-κB function in the nervous system. Front Immunol 10(MAY): 1043. 10.3389/fimmu.2019.01043
- Camara AKS, Zhou Y, Wen P-C, Tajkhorshid E, and Kwok W-M (2017). Mitochondrial VDAC1: A Key Gatekeeper as Potential Therapeutic Target. Front Physiol 8(JUN). 10.3389/fphys.2017.00460
- Vyssokikh MY, and Brdiczka D (2003). The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim Pol 50(2): 389–404. 10.18388/abp.2003_3693
- Robey RB, and Hay N (2006). Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 25(34): 4683–4696. 10.1038/sj.onc.1209595
- Chiara F, Castellaro D, Marin O, Petronilli V, Brusilow WS, Juhaszova M, Sollott SJ, Forte M, Bernardi P, and Rasola A (2008). Hexokinase II detachment from mitochondria triggers apoptosis through the permeability transition pore independent of voltage-dependent anion channels. PLoS One 3(3): e1852. 10.1371/journal.pone.0001852
- Halestrap AP, Pereira GC, and Pasdois P (2015). The role of hexokinase in cardioprotection – Mechanism and potential for translation. Br J Pharmacol 172(8): 2085–2100. 10.1111/bph.12899
- Andrienko TN, Pasdois P, Pereira GC, Ovens MJ, and Halestrap AP (2017). The role of succinate and ROS in reperfusion injury – A critical appraisal. J Mol Cell Cardiol 110: 1–14. 10.1016/j.yjmcc.2017.06.016
- Vyssokikh MY, Holtze S, Averina OA, Lyamzaev KG, Panteleeva AA, Marey M V., Zinovkin RA, Severin FF, Skulachev M V., Fasel N, Hildebrandt TB, and Skulachev VP (2020). Mild depolarization of the inner mitochondrial membrane is a crucial component of an anti-aging program. Proc Natl Acad Sci 117(12):6491-6501. 10.1073/pnas.1916414117
- Amigoni L, Martegani E, and Colombo S (2013). Lack of HXK2 Induces Localization of Active Ras in Mitochondria and Triggers Apoptosis in the Yeast Saccharomyces cerevisiae. Oxid Med Cell Longev 2013: 678473. 10.1155/2013/678473
- Pellegrino-Coppola D, Claringbould A, Stutvoet M, Boomsma D, Ikram MA, Slagboom E, Westra H-J, and Franke L (2020). Correction for both common and rare cell types in blood is important to identify genes that correlate with age. bioRxiv [Preprint]. Posted May 30, 2020. 10.1101/2020.05.28.120600
–
ACKNOWLEDGMENTS
I would like to express my gratitude to Dr. Giulia Callegaro for the constructive discussions and for helping in the revision of the paper.
COPYRIGHT
© 2020

Regulation of the mitochondrial permeability transition pore and its effects on aging by Pellegrino-Coppola is licensed under a Creative Commons Attribution 4.0 International License.








