
Research Reports:
Microbial Cell, Vol. 1, No. 12, pp. 416 - 424; doi: 10.15698/mic2014.12.179
Loss of wobble uridine modification in tRNA anticodons interferes with TOR pathway signaling
1 Institut für Biologie, Abteilung Mikrobiologie, Universität Kassel, D-34132 Kassel, Germany.
2 Present address: Molecular Oncology Program, Spanish National Cancer Centre (CNIO), Melchor Fernandez Almagro 3, Madrid, Spain.
# These authors contributed equally to the study.
Keywords: Saccharomyces cerevisiae, TOR signaling, rapamycin, Gln3, NCR, Sit4, Elongator complex, tRNA anticodon modification, tRNase zymocin.
Abbreviations:
NCR - nitrogen catabolite repression,
PP2A - type 2A protein phosphatases,
PPIases - peptidyl-prolyl cis/trans-isomerases,
TOR - target of rapamycin.
Received originally: 31/07/2014 Received in revised form: 23/11/2014
Accepted: 26/11/2014
Published: 29/11/2014
Correspondence:
Raffael Schaffrath, Institut für Biologie, Abteilung Mikrobiologie, Universität Kassel, Heinrich-Plett-Str. 40; D-34132 Kassel, Germany schaffrath@uni-kassel.de
Conflict of interest statement:
The authors declare there is no conflict of interest.
Please cite this article as: Viktor Scheidt, André Jüdes, Christian Bär, Roland Klassen and Raffael Schaffrath (2014). Loss of wobble uridine modification in tRNA anticodons interferes with TOR pathway signaling. Microbial Cell 1(12): 416-424.
Abstract
Previous work in yeast has suggested that modification of tRNAs, in particular uridine bases in the anticodon wobble position (U34), is linked to TOR (target of rapamycin) signaling. Hence, U34 modification mutants were found to be hypersensitive to TOR inhibition by rapamycin. To study whether this involves inappropriate TOR signaling, we examined interaction between mutations in TOR pathway genes (tip41∆, sap190∆, ppm1∆, rrd1∆) and U34 modification defects (elp3∆, kti12∆, urm1∆, ncs2∆) and found the rapamycin hypersensitivity in the latter is epistatic to drug resistance of the former. Epistasis, however, is abolished in tandem with a gln3∆ deletion, which inactivates transcription factor Gln3 required for TOR-sensitive activation of NCR (nitrogen catabolite repression) genes. In line with nuclear import of Gln3 being under control of TOR and dephosphorylation by the Sit4 phosphatase, we identify novel TOR-sensitive sit4 mutations that confer rapamycin resistance and importantly, mislocalise Gln3 when TOR is inhibited. This is similar to gln3∆ cells, which abolish the rapamycin hypersensitivity of U34 modification mutants, and suggests TOR deregulation due to tRNA undermodification operates through Gln3. In line with this, loss of U34 modifications (elp3∆, urm1∆) enhances nuclear import of and NCR gene activation (MEP2, GAP1) by Gln3 when TOR activity is low. Strikingly, this stimulatory effect onto Gln3 is suppressed by overexpression of tRNAs that usually carry the U34 modifications. Collectively, our data suggest that proper TOR signaling requires intact tRNA modifications and that loss of U34 modifications impinges on the TOR-sensitive NCR branch via Gln3 misregulation.
INTRODUCTION
While cell growth and proliferation are typically characterized by active de novo protein synthesis, cell quiescence goes along with translational downregulation. So, translational activity and metabolic cycling need to be tightly regulated in response to growth signals [1]. In eukaryotes including budding yeast, this is coordinated by the nutrient-sensitive TOR (target of rapamycin) kinase pathway, which among others promotes the biogenesis of components essential for translation, i.e. ribosomal proteins, rRNAs and tRNAs [1][2]. During maturation, tRNAs undergo many posttranscriptional modifications which appear to occupy roles in TOR-dependent processes [3][4][5]. In line with this notion are TOR-indicative defects in filamentous growth and metabolic cycling as well as GAAC (general amino acid control) response signatures typical of tRNA modification mutants including those that fail to form 5-methoxycarbonylmethyl-2-thiouridine (mcm5s2U) onto anticodon wobble uridines (U34) [5][6][7]. The mcm5s2U34 modification depends on two pathways one of which requires the Elongator complex (Elp1-Elp6) for mcm5 side chain formation while the second one (Uba4, Urm1, Ncs2/Ncs6) provides S-transfer for s2 thiolation [8][9][10]. In support of a link between TOR signaling and tRNA modification, Elongator and thiolation mutants are hypersensitive to TOR inhibition by caffeine and rapamycin [10][11][12][13][14].
–
When TOR activity is low due to nitrogen-starvation (or rapamycin treatment), type 2A protein phosphatases (PP2A) including Sit4 are typically freed from TOR control and dephosphorylate TOR pathway targets including Gln3 [1][2]. Gln3 is required for transcription of NCR (nitrogen catabolite repression) genes and its release from TOR phosphoinhibition by Sit4 dephosphorylation leads to its nuclear import and NCR gene activation [2][15]. Consistent with its TOR-sensitive role, loss of Gln3 causes rapamycin resistance. Gln3 activation also involves genes that encode regulators of PP2A including Sit4 (TAP42, TIP41, SAP190, PPM1/2, RRD1/2) and that, when mutated, protect against rapamycin [2][15][16][17][18]. Among these, the RRD1/2 products are PPIases (peptidyl-prolyl cis/trans-isomerases) which bind and activate PP2A enzymes through conformational changes thought to confer substrate specificity [19][20][21]. Consistent with such activator role for Sit4, rrd1/2∆ mutants accumulate Gln3 in its phosphorylated form [22]. Yet, sit4∆ cells are hypersensitive to rapamycin and this trait is unaltered in tandem with rrd1/2∆ null-alleles, which alone cause drug resistance [17][22][23]. This implies that the hypersensitivity of sit4∆ cells is independent of Rrd1/2 function. A TOR-independent role for Sit4 is indeed known, and was shown to confer growth inhibition by zymocin [17][24], a lethal tRNase toxin which kills cells by cleaving mcm5s2U34 modified anticodons(see above). Hence, tRNA modification defects typical of Elongator and sit4∆ mutants cause zymocin resistance [8][12][24][25][26]. sit4∆ mutants accumulate hyperphosphorylated Elongator forms demonstrating a link between Sit4 dephosphorylation and Elongator’s tRNA modification function [27][28]. This Sit4 role is independent of Rrd1/2 but requires the Sit4 partner proteins Sap185 and Sap190 [17][23][29], which is why a sap185∆190∆ double mutant copies defects typical of sit4∆ and Elongator mutants(i.e. Elongator hyperphosphorylation, loss of tRNA modification, zymocin resistance and rapamycin hypersensitivity) [25][26][27][28].
–
Here, we show that the rapamycin hypersensitivity of Elongator and U34 thiolation mutations (elp3∆, urm1∆), which are epistatic to TOR signaling mutations (tip41∆, sap190∆, ppm1∆, rrd1∆), is suppressed by overexpression of tRNAs known to undergo U34 modification and entirely abolished by a GLN3 deletion. This implies improper tRNA functioning due to loss of anticodon modifications interferes with the TOR signaling pathway through Gln3. Consistently, we can correlate mislocalisation of and upregulated NCR gene (MEP2, GAP1) activation by Gln3 with tRNA anticodon (U34) modification defects. This strongly suggests tRNA modifications are required for proper signaling into TOR-sensitive activation of NCR genes and regulation of Gln3.
RESULTS AND DISCUSSION
Previously, it was shown that inactivation of either Sit4 (sit4∆) or all four Sap proteins (sap4∆155∆185∆190∆) required for Sit4 function causes rapamycin hypersensitivity and that this trait is not altered in combination with an rrd1∆ null-allele [17][23]. Intriguingly, reintroduction of SAP190 into rrd1∆sap∆∆∆∆ cells reestablishes the drug resistance typical of rrd1∆ cells alone suggesting the trait depends on Sit4 and Sap190 [17]. This is similar to Sit4 dependent Elongator dephosphorylation, which requires Sap185 and Sap190 and promotes Elongator’s tRNA modification function [25][27][28]. Elongator dephosphorylation can be suppressed by high dosage of KTI12, a gene coding for a potential regulator of Elongator and Sit4 [12][27]. Strikingly, multicopy KTI12 gene dosage was also found to confer rapamycin sensitivity in an rrd1∆ background (Supplemental Figure 1). With Kti12 being intimately linked to Elongator [8][12][30][31], we observed that both Elongator and KTI12 gene deletions (elp3∆, kti12∆) phenocopied each other and caused rrd1∆ cells to become rapamycin sensitive (Figure 1A). Besides Elongator function, formation of the mcm5s2U34 modification also requires components of the U34 thiolation pathway (i.e. Urm1, Uba4 and Ncs2/Ncs6) [8][9][10]. When we combined sulfur transfer defects (urm1∆, ncs2∆) with an rrd1∆ allele, the resulting drug sensitivity basically copied the above epistasis seen with Elongator and kti12 mutations (Figure 1A). Similarly, on studying growth inhibition by caffeine, a TOR inhibitor drug distinct from rapamycin [32], we again found epistasis between U34 modification defects and an rrd1∆ null-allele confirming that the genetic interactions seen were specific to the TOR pathway (Supplemental Figure 2).
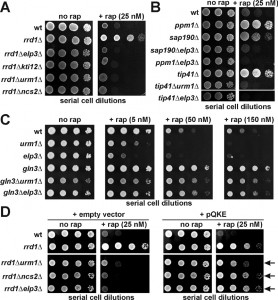 | FIGURE 1: Genetic interaction between mutations in TOR signaling and tRNA modification pathways. (A) Rapamycin hypersensitivity due to loss of tRNA modification in Elongator and U34 thiolation mutants is epistatic over TOR signaling mutant rrd1∆. (B) Other mutations in TOR pathway signaling genes cannot alter the rapamycin sensitive phenotype of Elongator or U34 thiolation mutants. (C) Hypersensitivity of U34 modification mutants to TOR inhibition by rapamycin requires Gln3 function. (D) Overexpression of tRNA species (tRNAGln, tRNALys and tRNAGlu) known to undergo U34 anticodon modification suppresses the rapamycin sensitivity of rrd1∆ cells carrying tRNA modification defects and reinstates drug tolerance. Shown are drug responses with vector controls (+ empty vector: left panels) or in response to tRNA overexpression from multicopy plasmid (+ pQKE: right panels) carrying tRNAGln (Q) tRNALys (K) and tRNAGlu (E) genes. Phenotypic suppression is indicated by arrows. In (A-D), ten-fold serial dilutions of yeast tester strains with genetic backgrounds as indicated were spotted onto YPD media containing no drug (no rap) or various doses (5, 20, 25, 50 and 150 nM) of rapamycin (+ rap) and grown for 3-5 days at 30°C. Lack of growth indicates rapamycin sensitive or hypersensitive responses while growth in the presence of the TOR inhibitor drug equals rapamycin resistance. |
Next, we included TOR signaling mutants with defects in the Gln3 pathway (sap190Δ, tip41Δ or ppm1Δ) [15][18] into our assays. We found that on their own, they copied the rapamycin resistance of rrd1∆ cells, but in tandem with an elp3Δ mutation, they also failed to counter the drug sensitivity typical of Elongator mutants (Figure 1B). Consistent with our observation that the drug sensitivity of thiolation defects was independent of rrd1Δ (Figure 1A), we observed that the rapamycin hypersensitivity of an urm1Δ mutant was not altered in tandem with a tip41Δ null-allele, which alone is a potent rapamycin suppressor (Figure 1B). Taken together, our data indicate that TOR-related phenotypes (rapamycin and caffeine hypersensitivity), which result from loss of U34 modifications in Elongator and thiolation mutants, are not modulated by Gln3 pathway gene mutations that cause protection against TOR inhibitor drugs.
–
Gln3 is a TOR-sensitive transcription factor necessary for NCR gene activation [33]. We found that the rapamycin resistance of a gln3Δ mutant was hardly altered in tandem with either elp3Δ or urm1Δ null-alleles (Figure 1C). So in striking contrast to the above TOR signaling mutants, the drug phenotype of gln3Δ cells is insensitive to tRNA modification defects indicating the rapamycin hypersensitivity of elp3Δ and urm1Δ mutants requires Gln3 function. Activity of Gln3 is largely regulated at the level of nucleo-cytoplasmic shuttling and cytosolic localization involves phosphoinhibition of Gln3 by TOR and interaction with Ure2 for cytosolic retention [34]. Consistently, inactivation of Ure2 in ure2Δ cells leads to nuclear import of Gln3, NCR gene activation and rapamycin hypersensitivity [34]. Having found that Gln3 is critical for the rapamycin hypersensitivity of U34 modification mutants, we asked if this trait may involve URE2 function. On comparing rapamycin phenotypes between ure2Δ and urm1Δ single as well as ure2Δurm1Δ double mutants, we observed a hypersensitive response of the latter to the TOR inhibitor drug (Supplemental Figure 3). Such phenotype suggests that in combination, defects in Ure2 and U34 modification are additive and result in enhanced sensitivity to TOR inhibition by rapamycin.
–
Studies in yeast have shown that U34 tRNA modification defects and associated phenotypes are suppressible by overexpressing tRNAs that would normally undergo the U34 anticodon modifications. Therefore, we repeated the above assays in the absence (empty vector) and presence of a multicopy plasmid (pQKE) that allows for overexpression of tRNAs (tRNAGln [Q], tRNALys [K] and tRNAGlu [E]) known to be mcm5s2U34 modified in wild-type cells (see above). As illustrated in Figure 1D, higher-than-normal levels of these tRNA species had efficient suppressor function and reconferred a rapamycin tolerant trait to rrd1∆elp3∆, rrd1∆urm1∆ and rrd1∆ncs2∆ double mutants that almost compares to the drug tolerance of an rrd1∆ single mutant alone. Similarly, we found that the caffeine sensitive phenotype typical of the rrd1∆elp3∆ double mutant was suppressed by tRNA overexpression (Supplemental Figure 4) to confer a drug tolerant trait resembling the rrd1∆ mutant alone. Taken together, these suppression data strongly suggest that the effect U34 modification defects obviously have on TOR pathway mutants including rrd1∆ cells can be ascribed to loss of Elongator’s tRNA modification function (elp3∆) and deficient thiolation (urm1∆, ncs2∆). So, proper tRNA anticodon (U34) modification and translation-related tRNA functions that are associated with it are apparently required for intact TOR pathway signaling.
–
With Gln3 localisation involving dephosphorylation by Sit4, we next asked whether TOR-dependent roles of this multifunctional PP2A phosphatase could be distinguished from TOR-independent ones. MammalianPP2A phosphatase activator (PTPA) and yeast Rrd1/2 are related PPIases with roles in phosphatase regulation [18][19][20][21][22][35]. Isomerization of human PP2A by PTPA involves a proline residue (P190) [19] which in yeast Sit4 highly likely aligns to P187. To study the significance of P187, substitutions (P187A/F) were generated and analysed in zymocin and rapamycin assays indicative for TOR-independent and TOR-dependent Sit4 functions [12][17][18]. As for the zymocin assays, a SIT4 wild-type allele, the substitutions (P187A/F) and empty vector were co-transformed into a sit4Δ reporter strain with pHMS14. pHMS14 allows for galactose-inducible expression of the tRNase γ-toxin subunit of zymocin, which in the presence of active Sit4 (and Elongator’s intact tRNA modification function) cleaves anticodons and becomes lethal [25][36][37]. Upon galactose induction, sit4Δ cells (with empty vector) survived tRNase expression. The SIT4 wild-type cells and both substitution (P187A/F) mutants, however, became killed (Figure 2A) indicating that P187 is dispensable for the role Sit4 plays in zymocin inhibition and Elongator’s tRNA modification function. In contrast, the rapamycin assays (Figure 2B) show that, similar to rrd1Δ cells, the sit4 mutants (P187A/F) are resistant against TOR inhibition by the drug. Based on these novel differential sit4 phenotypes, the substitutions (P187A/F) thus separate TOR-independent (zymocin action) from TOR-dependent (rapamycin inhibition) Sit4 functions. Remarkably, the novel sit4 mutations suppressed the rapamycin hypersensitivity of elp3Δ cells (Figure 2C). This is similar to the gln3Δ scenario (Figure 1C) and suggests that in the P187A/F mutants, non-functional Gln3 may be responsible for this elp3Δ suppressor effect. To address this issue in more detail, we found that in relation to SIT4 wild-type cells, the P187A/F mutants indeed mislocalised a GFP-Gln3 reporter and failed to shuttle the transcription factor into the nucleus following TOR inhibition by rapamycin (Figure 2D). So, cytosolic accumulation of GFP-Gln3 under conditions of TOR inactivation is in good agreement with our data showing that the P187A/F mutants evoke rapamycin resistance (Figure 2B), a trait indicative for Gln3 inactivation and copied by loss of Gln3 function in the gln3Δ null-mutant (Figure 1C).
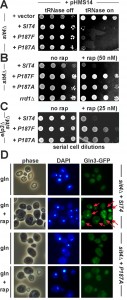 | FIGURE 2: Substitutions of Sit4 proline residue 187 identify novel sit4 mutants that separate TOR-dependent phosphatase functions from TOR-insensitive ones. (A) TOR-independent zymocin γ-toxin tRNase assay. The indicated sit4∆ backgrounds carrying empty vector, wild-type SIT4 and the P187F/A substitution alleles were transformed with the GAL1::γ-toxin expression vector (pHMS14) [12] and spotted onto glucose repressing (tRNase off) or galactose inducing (tRNase on) media. Growth was for 3 days at 30°C. Resistance or sensitivity towards tRNase toxicity is distinguished by growth or lack of growth, respectively. (B) TOR-sensitive rapamycin phenotype. Ten-fold serial cell dilutions of a TOR signaling mutant (rrd1∆) and sit4∆ cells with genetic backgrounds as indicated in (A) were spotted onto medium containing rapamycin (+ rap, 50 nM) or no drug (no rap). Lack of growth indicates drug sensitivity, growth equals rapamycin resistance. (C) The novel sit4 separation of function mutations suppress the sensitivity of Elongator mutants to rapamycin. A sit4∆elp3∆ double mutant with genetic backgrounds as indicated in (A) was grown on plates with no drug (no rap) or containing rapamycin (+ rap, 25 nM). (D) Gln3 mislocalises as a result of the P187 substitution and fails to be imported into the nucleus under conditions of TOR inhibition. Cells carrying pRS416-GFP-Gln3 were grown in minimal medium containing glutamine (gln) as the sole N-source with or without 10 nM rapamycin (rap) and images taken in phase contrast, DAPI- and GFP-fluorescence modes (phase, DAPI, Gln3-GFP). Arrows indicate GFP signals and foci that co-localise with DAPI-stained nuclei. |
Therefore, we next asked whether the loss of tRNA modification in elp3Δ or urm1Δ mutants may interfere with TOR signaling through effects onto Gln3. Using RT-PCR and qPCR, we studied TOR-sensitive transcription of NCR genes (GAP1, MEP2) by Gln3 in response to alterations of tRNA anticodon modification. The strains were either wild-type or deleted for ELP3 or URM1 or they carried sit4Δ (and sit4Δelp3Δ) or gln3Δ null-alleles in which phosphorylated Gln3 should remain cytosolic due to Sit4 defects (sit4Δ, sit4Δelp3Δ) or no NCR gene activation should occur in the first place (gln3Δ). Also, all strains were gat1Δ to eliminate gene activation by a transcription factor redundant to Gln3. As Gln3-independent controls, we monitored actin ACT1 (RT-PCR) and ALG9 (qPCR) gene transcription [38]. Under good nitrogen conditions (glutamine) and in the absence of rapamycin, we hardly detected any MEP2 and GAP1 activation by Gln3 in the tester strains (Figure 3A and Supplemental Figure 5A). This is consistent with phosphoinhibition by TOR and cytosolic localisation of Gln3 [22][33][34]. As expected, no MEP2 transcription was seen in gln3Δ, sit4Δ or sit4Δelp3Δ mutants (Figure 3A). Consistent with Gln3 being released from TOR control by rapamycin, addition of the drug triggered basal gene activation by Gln3 (Figure 3A and Supplemental Figure 5A). Strikingly, however, MEP2 and GAP1 gene transcription was significantly upregulated in elp3Δ or urm1Δ cells (Figure 3A and Supplemental Figure 5A) and based on validation by qPCR [38], MEP2 transcription increased roughly eight-fold (elp3Δ) and six-fold (urm1Δ) in relation to wild-type cells with proper tRNA modifications (Figure 3B). The finding that gene activation by Gln3 is significantly enhanced in the tRNA modification mutants is in line with our data showing that rapamycin hypersensitivity of elp3Δ or urm1Δ cells does require GLN3 function (Figure 1C). However, it is noteworthy that irrespective to TOR inhibition by rapamycin, even under conditions of TOR suppression by a poor nitrogen source (proline), constitutive Gln3-dependent transcription apparently differs between U34 modification mutants and wild-type cells (Supplemental Figure 5B).
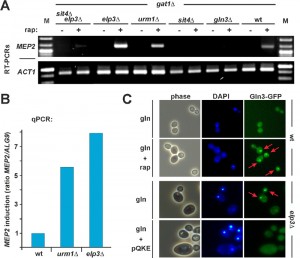 | FIGURE 3: Loss of U34 anticodon modification causes nuclear Gln3 mislocalisation and enhances TOR-sensitive gene activation by Gln3. (A, B) RT-PCR (A) and qPCR (B) reveal that the TOR-sensitive MEP2 gene transcription by Gln3 is enhanced in anticodon modification mutants that lack Elongator and U34 thiolation activities. Total RNA was isolated from the indicated strains cultivated with good nitrogen source (glutamine) supply in the absence (-) or presence (+) of 50 mM (rap) rapamycin. Following RT-PCR, the transcriptional induction of the MEP2 gene was analysed in comparison to actin (ACT1) transcription (A) and quantified by qPCR in relation to ALG9 transcription (B) (mean values of triplicates). (C) Gln3 mislocalises to the nucleus in an elp3∆ Elongator mutant, a property suppressible by tRNA overexpression. Wild-type cells carrying pRS416-GFP-Gln3 were grown in minimal medium containing glutamine (gln) as the sole N-source with or without 10 nM rapamycin (rap). For localization studies with the Elongator mutant in response to tRNA overexpression, elp3∆ cells carrying pRS416-GFP-Gln3 together with empty vector control or multicopy tRNA plasmid (pQKE) were used and images taken in phase contrast, DAPI- and GFP-fluorescence modes (phase, DAPI, Gln3-GFP). Arrows indicate nuclear localization of GFP-tagged Gln3. |
As for conditions of good nitrogen (glutamine) supply, our RT-PCR data show that enhanced MEP2 activation strictly depends on SIT4 function (Figure 3A), strongly suggesting that the transcription activation effects of U34 modification defects on Gln3 operate through the Sit4 phosphatase and require Gln3 dephosphorylation for nuclear localisation. Consistent with this notion, we observed that in the absence of TOR inhibition by rapamycin and in drastic contrast to wild-type cells, GFP-tagged Gln3 was significantly mislocalised in cells lacking the ELP3 gene and accumulated in the nucleus (Figure 3C). Equally important and consistent with our phenotypic suppression data above (Figure 1D and Supplemental Figure 4) was our finding that Gln3 mislocalisation in the Elongator mutant could be efficiently suppressed by overexpressing tRNAs (tRNAGln, tRNALys and tRNAGlu) usually carrying U34 anticodon modifications (Figure 3C). This suggests it is a tRNA-related function and/or process that, when impaired or deficient due to loss of Elongator’s tRNA modification function, affects proper Gln3 localisation and subsequent transcriptional activation. Whether misregulated Gln3 localization accounts for the activated NCR response signature in the Elongator mutant is an attractive option since it entirely consists with a previous report from 2007 [39] that placed Urm1 upstream of Gln3 and also showed Gln3 mislocalisation in an urm1Δ mutant, which by then was solely thought to be deficient in urmylation (a ubiquitin-like protein conjugation pathway) [39]. However, with the recently advanced evidence showing that Urm1 has dual roles in protein urmylation and tRNA anticodon (U34) thiolation [8][9][10], Gln3 mislocalization shared between elp3Δ (Figure 3C) and urm1Δ [39] cells further supports our notion that it is loss of tRNA anticodon modification due to Elongator inactivation or U34 thiolation defects (rather than protein urmylation) which enhances NCR gene activation by Gln3, particularly under conditions of low TOR activity (Figure 3A and Supplemental Figure 5A).
–
Our observation that improper tRNA modification deregulates transcription factor Gln3, is very reminiscent of two previous reports showing that Elongator-linked tRNA modification defects as well as KEOPS mutants with inappropriate N6-threonylcarbamoyl adenosine (t6A) modification in the anticodon stem loop can also misregulate transcription factor Gcn4 and the GAAC response associated with it [5][6]. Intriguingly, this is in line with another study showing that a sap185∆190∆ mutant which lacks the Sit4 partner proteins, Sap185 and Sap190, caused GCN4 induction, too [23]. This is significant since sap185∆190∆ cells copy defects and phenotypes typical of sit4∆ and Elongator mutants including loss of tRNA modification and rapamycin hypersensitivity [23][27][28][29]. Since protein dephosphorylation by Sit4 not only regulates Elongator’s tRNA modification function [27][28][29] but also operates on the two TOR pathway branches, NCR (Gln3) and GAAC (Gcn4) [15], it will be very important to study how the loss of tRNA modifications elicits Gln3 and Gcn4 response signatures at the molecular level. In yeast, almost all Elongator-linked phenotypes studied are rescued or at least partially suppressed by overexpressing tRNAs whose anticodons would normally carry the mcm5s2U34 modifications [10][40][41][42]. Consistent with this, we found that overexpressing tRNAGln, tRNALys and tRNAGlu is sufficient to suppress phenotypes (Figure 1D and Supplemental Figure 4) and properties that are typical of U34 modification mutants including Gln3 mislocalisation (Figure 3C). This strongly suggests that a translational defect in these tRNA modification mutants operates in deregulated TOR signaling. In support of this, a recent study has shown that U34 modifications indeed promote tRNA decoding functions during mRNA translation elongation [42].
–
Collectively, our data suggest that proper tRNA modifications play a positive role in the TOR signaling pathway. As a result of loss of anticodon modification, U34-minus cells undergo TOR suppression and enhance TOR-sensitive gene activation by Gln3 (NCR). Whether or not this operates upstream of TOR or outside the TOR network at a downstream level impinging on Gln3 activation, Ure2 interaction and/or nuclear import of Gln3 (Figure 4) is not entirely distinguishable from our preliminary evidence presented in this report. Alternatively, enhanced signaling in the U34 modification mutants may result from a combination of both options (Figure 4). In support of such scenario, our data show that drastic NCR gene activation by Gln3 in elp3Δ cells requires TOR inhibition by rapamycin for Gln3 mobilisation in concert with the Sit4 phosphatase, which (in addition to release of Gln3 from Ure2) further contributes to nuclear import of Gln3 and NCR gene transcription activation by Gln3. Whether the enhanced Gln3 activation (together with Gcn4 induction reported previously [5][6][43]) in tRNA modification mutants reflect cellular (stress) responses to overcome scenarios typically encountered upon TOR inhibition or suppression, i.e. poor nitrogen supply (NCR) or amino acid depletion (GAAC), are attractive hypotheses that need to be addressed in further studies. Given a report, however, that amino-acyl-tRNA synthetases have been found upregulated in tRNA modification mutants [5], tRNA charging defects, which may be associated with loss of U34 modifications, could potentially feed into TOR pathway signaling.
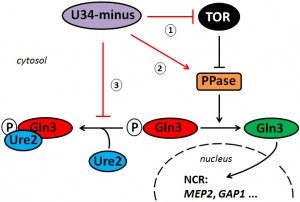 | FIGURE 4: Working model for interactions between the TOR pathway and tRNA modifications. Loss of anticodon wobble uridine (U34) modifications enhances rapamycin sensitivity and NCR gene activation by Gln3 suggesting U34-minus mutants dampen TOR signaling. Such buffer function may operate upstream of TOR (1) or outside from TOR (2, 3) affecting steps that counter TOR-sensitive Gln3 inhibition. The latter (2, 3) may involve dephosporylation by a phosphatase (PPase) such as Sit4 (2) and/or Gln3 release from Ure2 (3) for mobilization and nuclear import. Alternatively, the effects of inappropriate U34 modifications on Gln3 misregulation could result from a combination of these options (1 - 3). |
MATERIALS AND METHODS
General methods, yeast growth and strains
Routine yeast growth was in yeast extract, peptone, and dextrose (YPD) rich or synthetic complete (SC) minimal media [44]. For TOR modulation, growth media were supplemented with proline (poor N-source) or glutamine (good N-source). For testing the effect of TOR inhibitor drugs, 2.5 – 150 nM rapamycin (Calbiochem) or 2.5 – 15 mM, caffeine (Sigma) were added to YPD medium, and yeast growth was monitored for 3 – 5 days at 30°C. Yeast transformation used the PEG-lithium-acetate method [45] and tRNA overexpression involved multicopy plasmids pKQ/E carrying tRNALysUUU, tRNAGlnUUG and tRNAGluUUC genes [10]. To assess the response to lethal induction of zymocin’s tRNase subunit (γ-toxin), strains were transformed with pHMS14 [12], and growth was followed under conditions of tRNase expression (galactose) and repression (glucose). Substitutions of Sit4 proline residue 187 to either alanine or phenylalanine were generated using the two-step fusion PCR approach [46]. Mutants generated in this study by one-step PCR mediated gene deletion [47] derived from previously described strain backgrounds and used knock-out primers specific for ELP3, KTI12, URM1, NCS2 and GLN3 [12][13][17].
–
RT-PCR and qPCR methods
Total yeast RNA isolation and RT-PCR were carried out as previously described [36] with the following oligonucleotides ACT1-FW (5’-CTT CCG GTA GAA CTA CTG GT-3’); ACT1-RV (5’-CCT TAC GGA CAT CGA CAT CA-3’); GAP1-FW (5’-TCC CGC TTC GCT ACT GAT TG-3’); GAP1-RV (5’-GCA GAG TTA CCG ACA GAT AA-3’); MEP2-FW (5’-GGT ATG TTT GCC GCA GTC AC-3’) and MEP2-RV (5’-ACC ACC CAC ACC ATG GAT AG-3’). Real-time PCR used a Mastercycler (Eppendorf), the SensiFAST™ SYBR® No-ROX Kit (BIOLINE) and the ALG9 standardization protocol [38]. qPCR involved ALG9-qFW (5’-GTC ACG GAT AGT GGC TTT GG-3’); ALG9-qRV (5’-TGG CAG CAG GAA AGA ACT TG-3’); MEP2-qFW (5’-GTA TGT TTG CCG CAG TCA CC-3’) and MEP2-qRV (5’-CAG ACC CAG CAT GCA ATA GG-3’) oligonucleotides.
–
Cellular localization of Gln3-GFP
Yeast strains carrying pRS416-GFP-Gln3 [48][49] were grown in yeast nitrogen base media with glutamine as the sole N-source, left untreated or treated with 10 nM rapamycin for 20 min. Subsequently, cells were fixed by adding 3.7% formaldehyde directly to the medium and incubated for 10 min at room temperature and washed once with water. Cells were resuspended in water containing 1 µg•ml-1 4,6 diamidino-2-phenylindole (DAPI, Sigma, Germany). Following washing with water, cells were analyzed using an Olympus BX53 microscope with appropriate filters for DAPI and GFP fluorescence. Images were captured using the CellSens 1.6 software package (Olympus).
References
- J.R. Broach, "Nutritional Control of Growth and Development in Yeast", Genetics, vol. 192, pp. 73-105, 2012. http://dx.doi.org/10.1534/genetics.111.135731
- R. Loewith, and M.N. Hall, "Target of Rapamycin (TOR) in Nutrient Signaling and Growth Control", Genetics, vol. 189, pp. 1177-1201, 2011. http://dx.doi.org/10.1534/genetics.111.133363
- B. El Yacoubi, M. Bailly, and V. de Crécy-Lagard, "Biosynthesis and Function of Posttranscriptional Modifications of Transfer RNAs", Annual Review of Genetics, vol. 46, pp. 69-95, 2012. http://dx.doi.org/10.1146/annurev-genet-110711-155641
- P.J. Cullen, and G.F. Sprague, "The Regulation of Filamentous Growth in Yeast", Genetics, vol. 190, pp. 23-49, 2012. http://dx.doi.org/10.1534/genetics.111.127456
- B. Zinshteyn, and W.V. Gilbert, "Loss of a Conserved tRNA Anticodon Modification Perturbs Cellular Signaling", PLoS Genetics, vol. 9, pp. e1003675, 2013. http://dx.doi.org/10.1371/journal.pgen.1003675
- M. Daugeron, T.L. Lenstra, M. Frizzarin, B. El Yacoubi, X. Liu, A. Baudin-Baillieu, P. Lijnzaad, L. Decourty, C. Saveanu, A. Jacquier, F.C.P. Holstege, V. de Crécy-Lagard, H. van Tilbeurgh, and D. Libri, "Gcn4 misregulation reveals a direct role for the evolutionary conserved EKC/KEOPS in the t6A modification of tRNAs", Nucleic Acids Research, vol. 39, pp. 6148-6160, 2011. http://dx.doi.org/10.1093/nar/gkr178
- S. Laxman, B. Sutter, X. Wu, S. Kumar, X. Guo, D. Trudgian, H. Mirzaei, and B. Tu, "Sulfur Amino Acids Regulate Translational Capacity and Metabolic Homeostasis through Modulation of tRNA Thiolation", Cell, vol. 154, pp. 416-429, 2013. http://dx.doi.org/10.1016/j.cell.2013.06.043
- B. HUANG, M.J. JOHANSSON, and A.S. BYSTRÖM, "An early step in wobble uridine tRNA modification requires the Elongator complex", RNA, vol. 11, pp. 424-436, 2005. http://dx.doi.org/10.1261/rna.7247705
- A. Noma, Y. Sakaguchi, and T. Suzuki, "Mechanistic characterization of the sulfur-relay system for eukaryotic 2-thiouridine biogenesis at tRNA wobble positions", Nucleic Acids Research, vol. 37, pp. 1335-1352, 2009. http://dx.doi.org/10.1093/nar/gkn1023
- S. Leidel, P.G.A. Pedrioli, T. Bucher, R. Brost, M. Costanzo, A. Schmidt, R. Aebersold, C. Boone, K. Hofmann, and M. Peter, "Ubiquitin-related modifier Urm1 acts as a sulphur carrier in thiolation of eukaryotic transfer RNA", Nature, vol. 458, pp. 228-232, 2009. http://dx.doi.org/10.1038/nature07643
- T. Chan, J. Carvalho, L. Riles, and X.F.S. Zheng, "A chemical genomics approach toward understanding the global functions of the target of rapamycin protein (TOR)", Proceedings of the National Academy of Sciences, vol. 97, pp. 13227-13232, 2000. http://dx.doi.org/10.1073/pnas.240444197
- F. Frohloff, "Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin", The EMBO Journal, vol. 20, pp. 1993-2003, 2001. http://dx.doi.org/10.1093/emboj/20.8.1993
- L. Fichtner, D. Jablonowski, A. Schierhorn, H.K. Kitamoto, M.J.R. Stark, and R. Schaffrath, "Elongator's toxin‐target (TOT) function is nuclear localization sequence dependent and suppressed by post‐translational modification", Molecular Microbiology, vol. 49, pp. 1297-1307, 2003. http://dx.doi.org/10.1046/j.1365-2958.2003.03632.x
- A.S. Goehring, D.M. Rivers, and G.F. Sprague, "Urmylation: A Ubiquitin-like Pathway that Functions during Invasive Growth and Budding in Yeast", Molecular Biology of the Cell, vol. 14, pp. 4329-4341, 2003. http://dx.doi.org/10.1091/mbc.E03-02-0079
- K. Düvel, and J.R. Broach, "The role of phosphatases in TOR signaling in yeast.", Current topics in microbiology and immunology, 2004. http://www.ncbi.nlm.nih.gov/pubmed/14560949
- T.K. Neklesa, and R.W. Davis, "Superoxide anions regulate TORC1 and its ability to bind Fpr1:rapamycin complex", Proceedings of the National Academy of Sciences, vol. 105, pp. 15166-15171, 2008. http://dx.doi.org/10.1073/pnas.0807712105
- D. Jablonowski, J. Täubert, C. Bär, M.J.R. Stark, and R. Schaffrath, "Distinct Subsets of Sit4 Holophosphatases Are Required for Inhibition ofSaccharomyces cerevisiaeGrowth by Rapamycin and Zymocin", Eukaryotic Cell, vol. 8, pp. 1637-1647, 2009. http://dx.doi.org/10.1128/EC.00205-09
- B. Rempola, A. Kaniak, A. Migdalski, J. Rytka, P.P. Slonimski, and J.P. di Rago, "Functional analysis of RRD1 (YIL153w) and RRD2 (YPL152w), which encode two putative activators of the phosphotyrosyl phosphatase activity of PP2A in Saccharomyces cerevisiae.", Molecular & general genetics : MGG, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10660069
- J. Jordens, V. Janssens, S. Longin, I. Stevens, E. Martens, G. Bultynck, Y. Engelborghs, E. Lescrinier, E. Waelkens, J. Goris, and C. Van Hoof, "The Protein Phosphatase 2A Phosphatase Activator Is a Novel Peptidyl-Prolyl cis/trans-Isomerase", Journal of Biological Chemistry, vol. 281, pp. 6349-6357, 2006. http://dx.doi.org/10.1074/jbc.M507760200
- N. Leulliot, G. Vicentini, J. Jordens, S. Quevillon-Cheruel, M. Schiltz, D. Barford, H. Van Tilbeurgh, and J. Goris, "Crystal Structure of the PP2A Phosphatase Activator: Implications for Its PP2A-Specific PPIase Activity", Molecular Cell, vol. 23, pp. 413-424, 2006. http://dx.doi.org/10.1016/j.molcel.2006.07.008
- H. Hombauer, D. Weismann, I. Mudrak, C. Stanzel, T. Fellner, D.H. Lackner, and E. Ogris, "Generation of Active Protein Phosphatase 2A Is Coupled to Holoenzyme Assembly", PLoS Biology, vol. 5, pp. e155, 2007. http://dx.doi.org/10.1371/journal.pbio.0050155
- Y. Zheng, and Y. Jiang, "The Yeast Phosphotyrosyl Phosphatase Activator Is Part of the Tap42–Phosphatase Complexes", Molecular Biology of the Cell, vol. 16, pp. 2119-2127, 2005. http://dx.doi.org/10.1091/mbc.E04-09-0797
- J.R. Rohde, S. Campbell, S.A. Zurita-Martinez, N.S. Cutler, M. Ashe, and M.E. Cardenas, "TOR Controls Transcriptional and Translational Programs via Sap-Sit4 Protein Phosphatase Signaling Effectors", Molecular and Cellular Biology, vol. 24, pp. 8332-8341, 2004. http://dx.doi.org/10.1128/MCB.24.19.8332-8341.2004
- D. Jablonowski, and R. Schaffrath, "Zymocin, a composite chitinase and tRNase killer toxin from yeast", Biochemical Society Transactions, vol. 35, pp. 1533-1537, 2007. http://dx.doi.org/10.1042/BST0351533
- D. Jablonowski, A.R. Butler, L. Fichtner, D. Gardiner, R. Schaffrath, and M.J. Stark, "Sit4p protein phosphatase is required for sensitivity of Saccharomyces cerevisiae to Kluyveromyces lactis zymocin.", Genetics, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11779790
- B. Huang, J. Lu, and A.S. Byström, "A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae", RNA, vol. 14, pp. 2183-2194, 2008. http://dx.doi.org/10.1261/rna.1184108
- D. Jablonowski, L. Fichtner, M.J. Stark, and R. Schaffrath, "The Yeast Elongator Histone Acetylase Requires Sit4-dependent Dephosphorylation for Toxin-Target Capacity", Molecular Biology of the Cell, vol. 15, pp. 1459-1469, 2004. http://dx.doi.org/10.1091/mbc.E03-10-0750
- C. Mehlgarten, D. Jablonowski, K.D. Breunig, M.J.R. Stark, and R. Schaffrath, "Elongator function depends on antagonistic regulation by casein kinase Hrr25 and protein phosphatase Sit4", Molecular Microbiology, vol. 73, pp. 869-881, 2009. http://dx.doi.org/10.1111/j.1365-2958.2009.06811.x
- M.M. Luke, F. Della Seta, C.J. Di Como, H. Sugimoto, R. Kobayashi, and K.T. Arndt, "The SAP, a new family of proteins, associate and function positively with the SIT4 phosphatase.", Molecular and cellular biology, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8649382
- L. Fichtner, F. Frohloff, K. Bürkner, M. Larsen, K.D. Breunig, and R. Schaffrath, "Molecular analysis of KTI12/TOT4, a Saccharomyces cerevisiae gene required for Kluyveromyces lactis zymocin action", Molecular Microbiology, vol. 43, pp. 783-791, 2002. http://dx.doi.org/10.1046/j.1365-2958.2002.02794.x
- C. Mehlgarten, D. Jablonowski, U. Wrackmeyer, S. Tschitschmann, D. Sondermann, G. Jäger, Z. Gong, A.S. Byström, R. Schaffrath, and K.D. Breunig, "Elongator function in tRNA wobble uridine modification is conserved between yeast and plants", Molecular Microbiology, vol. 76, pp. 1082-1094, 2010. http://dx.doi.org/10.1111/j.1365-2958.2010.07163.x
- A. Reinke, J.C. Chen, S. Aronova, and T. Powers, "Caffeine Targets TOR Complex I and Provides Evidence for a Regulatory Link between the FRB and Kinase Domains of Tor1p", Journal of Biological Chemistry, vol. 281, pp. 31616-31626, 2006. http://dx.doi.org/10.1074/jbc.M603107200
- T. Beck, and M.N. Hall, "The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors.", Nature, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10604478
- P.G. Bertram, J.H. Choi, J. Carvalho, W. Ai, C. Zeng, T. Chan, and X.S. Zheng, "Tripartite Regulation of Gln3p by TOR, Ure2p, and Phosphatases", Journal of Biological Chemistry, vol. 275, pp. 35727-35733, 2000. http://dx.doi.org/10.1074/jbc.M004235200
- C. Van HOOF, E. MARTENS, S. LONGIN, J. JORDENS, I. STEVENS, V. JANSSENS, and J. GORIS, "Specific interactions of PP2A and PP2A-like phosphatases with the yeast PTPA homologues, Ypa1 and Ypa2", Biochemical Journal, vol. 386, pp. 93-102, 2005. http://dx.doi.org/10.1042/BJ20040887
- D. Jablonowski, S. Zink, C. Mehlgarten, G. Daum, and R. Schaffrath, "tRNAGlu wobble uridine methylation by Trm9 identifies Elongator's key role for zymocin‐induced cell death in yeast", Molecular Microbiology, vol. 59, pp. 677-688, 2005. http://dx.doi.org/10.1111/j.1365-2958.2005.04972.x
- J. LU, B. HUANG, A. ESBERG, M.J. JOHANSSON, and A.S. BYSTRÖM, "The Kluyveromyces lactis γ-toxin targets tRNA anticodons", RNA, vol. 11, pp. 1648-1654, 2005. http://dx.doi.org/10.1261/rna.2172105
- M. Teste, M. Duquenne, J.M. François, and J. Parrou, "Validation of reference genes for quantitative expression analysis by real-time RT-PCR in Saccharomyces cerevisiae", BMC Molecular Biology, vol. 10, 2009. http://dx.doi.org/10.1186/1471-2199-10-99
- M. Rubio-Texeira, "Urmylation controls Nil1p and Gln3p‐dependent expression of nitrogen‐catabolite repressed genes in Saccharomyces cerevisiae", FEBS Letters, vol. 581, pp. 541-550, 2007. http://dx.doi.org/10.1016/j.febslet.2007.01.019
- A. Esberg, B. Huang, M.J. Johansson, and A.S. Byström, "Elevated Levels of Two tRNA Species Bypass the Requirement for Elongator Complex in Transcription and Exocytosis", Molecular Cell, vol. 24, pp. 139-148, 2006. http://dx.doi.org/10.1016/j.molcel.2006.07.031
- C. Chen, B. Huang, M. Eliasson, P. Rydén, and A.S. Byström, "Elongator Complex Influences Telomeric Gene Silencing and DNA Damage Response by Its Role in Wobble Uridine tRNA Modification", PLoS Genetics, vol. 7, pp. e1002258, 2011. http://dx.doi.org/10.1371/journal.pgen.1002258
- V.A.N. Rezgui, K. Tyagi, N. Ranjan, A.L. Konevega, J. Mittelstaet, M.V. Rodnina, M. Peter, and P.G.A. Pedrioli, "tRNA tK UUU , tQ UUG , and tE UUC wobble position modifications fine-tune protein translation by promoting ribosome A-site binding", Proceedings of the National Academy of Sciences, vol. 110, pp. 12289-12294, 2013. http://dx.doi.org/10.1073/pnas.1300781110
- N.J. Krogan, and J.F. Greenblatt, "Characterization of a Six-Subunit Holo-Elongator Complex Required for the Regulated Expression of a Group of Genes in Saccharomyces cerevisiae", Molecular and Cellular Biology, vol. 21, pp. 8203-8212, 2001. http://dx.doi.org/10.1128/MCB.21.23.8203-8212.2001
- F. Sherman, "Getting started with yeast.", Methods in enzymology, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12073320
- D. Gietz, A.S. Jean, R.A. Woods, and R.H. Schiestl, "Improved method for high efficiency transformation of intact yeast cells", Nucleic Acids Research, vol. 20, pp. 1425-1425, 1992. http://dx.doi.org/10.1093/nar/20.6.1425
- O. Hobert, "PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegans.", BioTechniques, 2002. http://www.ncbi.nlm.nih.gov/pubmed/11962590
- C. Janke, M.M. Magiera, N. Rathfelder, C. Taxis, S. Reber, H. Maekawa, A. Moreno‐Borchart, G. Doenges, E. Schwob, E. Schiebel, and M. Knop, "A versatile toolbox for PCR‐based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes", Yeast, vol. 21, pp. 947-962, 2004. http://dx.doi.org/10.1002/yea.1142
- S. Giannattasio, Z. Liu, J. Thornton, and R.A. Butow, "Retrograde Response to Mitochondrial Dysfunction Is Separable from TOR1/2 Regulation of Retrograde Gene Expression", Journal of Biological Chemistry, vol. 280, pp. 42528-42535, 2005. http://dx.doi.org/10.1074/jbc.M509187200
- I. Georis, J.J. Tate, T.G. Cooper, and E. Dubois, "Nitrogen-responsive Regulation of GATA Protein Family Activators Gln3 and Gat1 Occurs by Two Distinct Pathways, One Inhibited by Rapamycin and the Other by Methionine Sulfoximine", Journal of Biological Chemistry, vol. 286, pp. 44897-44912, 2011. http://dx.doi.org/10.1074/jbc.M111.290577
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We thank Drs S. Leidel and T. Cooper for tRNA overexpression and GFP-Gln3 localisation plasmids. We appreciate discussion with Dr T. Cooper and support by Zentrale Forschungsförderung (ZFF #1798), Universität Kassel, Germany, and Deutsche Forschungsgemeinschaft (SCHA750/15 & SCHA750/18), Bonn, Germany.
COPYRIGHT
© 2014

Loss of wobble uridine modification in tRNA anticodons interferes with TOR pathway signaling by Viktor Scheidt et al. is licensed under a Creative Commons Attribution 4.0 International License.








