
Reviews:
Microbial Cell, Vol. 5, No. 7, pp. 300 - 326; doi: 10.15698/mic2018.07.638
Methodologies for in vitro and in vivo evaluation of efficacy of antifungal and antibiofilm agents and surface coatings against fungal biofilms

1 VIB-KU Leuven Center for Microbiology, Leuven, Belgium.
2 KU Leuven Laboratory of Molecular Cell Biology, Leuven, Belgium.
3 University of Groningen, University Medical Center Groningen, Department of BioMedical Engineering, Groningen, The Netherlands.
4 Centre for Microbial and Plant Genetics, KU Leuven, Leuven, Belgium.
5 Department of Plant Systems Biology, VIB, Ghent, Belgium.
6 Department of Microbiology and Immunology, KU Leuven, Leuven, Belgium.
7 Clinical Department of Laboratory Medicine and National Reference Center for Mycosis, UZ Leuven, Belgium.
8 School of Molecular Cell Biology and Biotechnology, Faculty of Life Sciences, Tel Aviv University, Ramat Aviv, Israel.
9 Institut Pasteur, INRA, Unité Biologie et Pathogénicité Fongiques, Paris, France.
10 Department of Medical Microbiology and Immunology, University of Wisconsin-Madison, Madison, Wisconsin, USA.
11 Department of Medicine, University of Wisconsin-Madison, Madison, Wisconsin, USA.
12 Unit of Mycology, Statens Serum Institut, Copenhagen, Denmark.
13 Department of Clinical Microbiology, Rigshospitalet, Copenhagen, Denmark.
14 Department of Clinical Medicine, University of Copenhagen, Copenhagen, Denmark.
15 Leibniz Institute for Natural Product Research and Infection Biology – Hans Knoell Institute (HKI), Dept. Microbiology and Molecular Biology, Friedrich Schiller University Jena, Institute of Microbiology, Jena, Germany.
16 Department of Microbiology & Immunology, Georgetown University Medical Center, Washington DC, USA.
17 Severe Infection Research Group: Medical Research Institute La Fe (IISLaFe), Valencia, Spain.
18 Laboratory of Pharmaceutical Microbiology, Ghent University, Ghent, Belgium.
19 ESCMID Study Group for Biofilms, Switzerland.
20 Laboratory for Microbiology, Parasitology and Hygiene (LMPH), University of Antwerp, Belgium.
21 Department of Molecular Genetics, University of Toronto, Toronto, Ontario, Canada.
22 Department of Oral Biology, School of Dental Medicine, University at Buffalo, Buffalo, NY USA.
23 VCU Medical Center, Richmond, VA, USA.
24 Division of Infectious Diseases, Los Angeles Biomedical Research Institute at Harbor-UCLA Medical Center, Torrance, CA, USA.
25 Center for Medical Mycology, Department of Dermatology, University Hospitals Cleveland Medical Center and Case Western Reserve University, Cleveland, OH, USA.
26 MRC Centre for Medical Mycology, Institute of Medical Sciences, University of Aberdeen, Aberdeen, UK.
27 Biocenter – Division of Molecular Biology, Medical University Innsbruck, Innsbruck, Austria.
28 Department of Oncology and Diagnostic Sciences, School of Dentistry; Department of Microbiology and Immunology, School of Medicine, University of Maryland, Baltimore, USA.
29 National Infection Service, Public Health England, Mycology Reference Laboratory, Bristol, UK.
30 Centers for Disease Control and Prevention, Atlanta, GA, USA.
31 Department of Biology, South Texas Center for Emerging Infectious Diseases, The University of Texas at San Antonio, San Antonio, USA.
32 Department of Microbiology and Immunology, KU Leuven, Leuven, Belgium and Clinical Department of Haematology, UZ Leuven, Leuven, Belgium.
33 University of Wisconsin-Madison, Departments of Medicine and Medical Microbiology & Immunology, Madison, WI, USA.
34 Department of Molecular and Cell Biology, School of Natural Sciences, University of California, Merced, Merced, USA.
35 Departments of Pathology and Epidemiology, University of Iowa, Iowa, USA.
36 JMI Laboratories, North Liberty, Iowa, USA.
37 College of Medical, Veterinary and Life Sciences, University of Glasgow, UK.
38 Institute of Microbiology, University of Lausanne and University Hospital, CH-1011 Lausanne
39 Institute of Microbiology, Università Cattolica del Sacro Cuore, IRCCS-Fondazione Policlinico “Agostino Gemelli”, Rome, Italy.
40 Pharmacy Dpt, University Hospitals Leuven and Clinical Pharmacology and Pharmacotherapy, Dpt. of Pharmaceutical and Pharmacological Sciences, KU Leuven, Belgium.
41 Center of Expertise in Mycology Radboudumc/CWZ, Radboud University Medical Center, Nijmegen, the Netherlands (omit “Nijmegen” in Radboud University Medical Center).
42 MRC Centre for Medical Mycology, Aberdeen Fungal Group, University of Aberdeen, Foresterhill, Aberdeen, UK.
43 KU Leuven-University of Leuven, University Hospitals Leuven, Department of General Internal Medicine, Herestraat 49, B-3000 Leuven, Belgium.
44 Geffen School of Medicine at the University of California, Los Angeles, Divisions of Molecular Medicine & Infectious Diseases, Harbor-UCLA Medical Center, LABioMed at Harbor-UCLA Medical Center.
45 Department of Medical Microbiology, Amsterdam Infection and Immunity Institute, Academic Medical Center, University of Amsterdam, Netherlands.
‡ Equally contributing
Keywords: antifungal susceptibility testing, biofilm inhibition, biofilm eradication, antibiofilm material coating, in vivo models.
Received originally: 07/05/2018 Accepted: 24/05/2018
Published: 14/06/2018
Correspondence:
Karin Thevissen, CMPG, Kasteelpark Arenberg 20, B-3001 Heverlee, Belgium; karin.thevissen@kuleuven.be
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Patrick Van Dijck, Jelmer Sjollema, Bruno P.A. Cammue, Katrien Lagrou, Judith Berman, Christophe d’Enfert, David R. Andes, Maiken C. Arendrup, Axel A. Brakhage, Richard Calderone, Emilia Cantón, Tom Coenye, Paul Cos, Leah E. Cowen, Mira Edgerton, Ana Espinel-Ingroff, Scott G. Filler, Mahmoud Ghannoum, Neil A.R. Gow, Hubertus Haas, Mary-Ann Jabra-Rizk, Elizabeth M. Johnson, Shawn R. Lockhart, Jose L. Lopez-Ribot, Johan Maertens, Carol A. Munro, Jeniel E. Nett, Clarissa J. Nobile, Michael A. Pfaller, Gordon Ramage, Dominique Sanglard, Maurizio Sanguinetti, Isabel Spriet, Paul Verweij, Adilia Warris, Joost Wauters, Michael R. Yeaman, Sebastiaan A.J. Zaat, Karin Thevissen (2018). Methodologies for in vitro and in vivo evaluation of efficacy of antifungal and antibiofilm agents and surface coatings against fungal biofilms. Microbial Cell X(Y): in press.
Abstract
Unlike superficial fungal infections of the skin and nails, which are the most common fungal diseases in humans, invasive fungal infections carry high morbidity and mortality, particularly those associated with biofilm formation on indwelling medical devices. Therapeutic management of these complex diseases is often complicated by the rise in resistance to the commonly used antifungal agents. Therefore, the availability of accurate susceptibility testing methods for determining antifungal resistance, as well as discovery of novel antifungal and antibiofilm agents, are key priorities in medical mycology research. To direct advancements in this field, here we present an overview of the methods currently available for determining (i) the susceptibility or resistance of fungal isolates or biofilms to antifungal or antibiofilm compounds and compound combinations; (ii) the in vivo efficacy of antifungal and antibiofilm compounds and compound combinations; and (iii) the in vitro and in vivo performance of anti-infective coatings and materials to prevent fungal biofilm-based infections.
INTRODUCTION
Superficial fungal infections of the skin and nails are the most common fungal infections in humans and, although rarely invasive, they can be debilitating, persistent and impose substantial treatment costs [1]. In contrast, invasive fungal infections are life threatening, with a higher mortality rate per year than that by malaria, breast or prostate cancer [2]. More than 90% of all reported fungal-related deaths (about one million people every year) result from species that belong to one of four genera: Cryptococcus, Candida, Aspergillus and Pneumocystis [2][3].
–
The most important antifungal agents (antimycotics) clinically used for systemic infections can be subdivided into four main classes: azoles, polyenes, echinocandins and pyrimidine analogues (5-fluorocytosine). In addition, allylamines (terbinafine) are frequently used against superficial fungal infections [4]. The rise in azole resistance, echinocandin resistance and cross-resistance to at least 2 antifungal classes (multi-drug resistance: MDR) has been a worrisome trend, mainly in large tertiary and oncology centers. Overall, rates of antifungal resistance and MDR in Candida species and particularly in the emerging human pathogen C. glabrata are increasing [5]. More concerning, the newly identified Candida species C. auris has drawn considerable attention as this uncommon species is the first globally emerging fungal pathogen exhibiting MDR to the three major classes of antifungals (azoles, echinocandins and amphotericin B and its lipid formulations) and is characterized by a strong potential for nosocomial transmission [6]. In addition to Candida, azole resistance in Aspergillus fumigatus has been reported worldwide, and such resistant isolates can cause invasive infections with high mortality rates [7]. Alongside the serious issues presented by classical MDR, there is another important, but less appreciated problem with our current approach to antimicrobial therapy in general. Existing antimicrobial treatments are frequently associated with therapeutic failure even against infections caused by susceptible strains due to intrinsic mechanisms that protect the micro-organisms from the antimicrobial agents, such as the formation of drug-tolerant biofilms. Microbial biofilms consist of dense layers of microorganisms surrounded by an extracellular polymer matrix, which provides biofilm-embedded microorganisms with protection against antimicrobial agents. Most bacteria and fungi exist predominantly in such organized communities in nature and, according to a recent public announcement from the US NIH, biofilms are responsible for more than 80% of human soft- and hard-tissue infections [8]. Of more significance, microbial biofilms are thought to result in therapeutic failure and occurrence of resistance [9][10][11][12].
–
Therefore, the development of accurate susceptibility testing methods for detecting or excluding antifungal resistance, as well as discovery of novel antifungal and antibiofilm agents, are key priorities in medical mycology research. The term ‘antibiofilm agents’ relates to compounds that can inhibit biofilm formation and/or eradicate fungal cells in the biofilm.
–
To direct advancements in this field, we present in this review an overview of methods for use by investigators who aim to examine:
–
(i) susceptibility (and resistance) of fungal cultures or biofilms against antifungal or antibiofilm compounds and compound combinations;
–
(ii) in vivo efficacy of antifungal and antibiofilm compounds and compound combinations; and
–
(iii) in vitro and in vivo performance of anti-infective coatings and materials to prevent fungal biofilm-related infections.
–
Several of these topics are already covered in recent guideline-style based reviews [13][14][15][16]. We refer to these reviews in the relevant sections and summarize their most important recommendations and guidelines.
–
| The Minimum Inhibitory Concetration (MIC) is defined as the lowest concentration of an antimicrobial agent that inhibits microbial growth, as established by a standardized endpoint. Standardized MIC endpoints include partial inhibition relative to the growth control (50% inhibition of yeast growth as determined visually [CLSI] or spectrophotometrically [EUCAST] for azoles and echinocandins), complete inhibition (100%) of visual growth (CLSI) or >90% by spectrophotometer (EUCAST) as applied to amphotericin B. NOTE that MIC50, MIC90 and MIC100 should not be used in this context. |
| The MIC50 is the concentration of an antimicrobial agent at which 50 percent of the organisms tested are inhibited. The MIC90 is the concentration of an antimicrobial agent at which 90 percent of the organisms tested are inhibited. |
| The minimum effective concentration (MEC) is defined as the lowest concentration of an echinocandin that results in growth of filamentous fungi producing conspicuously aberrant growth. Aberrant growth of hyphae is defined as small, round, compact microcolonies compared with the matt of hyphal growth in the control well that does not contain an antifungal agent. |
| The minimum inhibitory concentration of a (novel) antimicrobial agent that inhibits the growth of fungi by 50% or 90%, respectively. |
| The minimum fungicidal/lethal concentration, defined as the minimum concentration of the antifungal drug resulting in a 99.9% reduction of fungal cell counts of the starting inoculum after a fixed time of incubation. |
| 50% and 90% effective concentrations and maximum effect, the minimum concentrations of the antifungal drug resulting in a net reduction in the number of CFU per milliliter from the starting inoculum by 50%, 90%, and 99.9% respectively. |
| The minimum concentration of an antifungal drug against sessile cultures (biofilm), in most cases based on a viability readout (e.g. an 80% reduction in the metabolic activity of the biofilm treated with the antifungal compared with the control well). |
| The minimum antibiofilm drug concentration resulting in a 2-fold inhibition of biofilm formation (based on readout relying on viability dyes or CFU counts). |
| The minimum antibiofilm drug concentration resulting in eradication of mature biofilms by 50% (based on readout relying on viability dyes or CFU counts). |
| The fractional inhibitory concentration index, determined as d1/(D1)p + d2/(D2)p, where d1 and d2 are the doses of compounds 1 and 2 in combination to result in a particular readout and (D1)p and (D2)p are the doses required for the two respective compounds alone to produce the same effect. An interaction is scored as indifferent/additive if 0.5<FICI<4; as antagonistic if FICI >4 or synergistic if FICI <0.5. |
METHODS FOR ANTIFUNGAL SUSCEPTIBILITY TESTING OF PLANKTONIC CULTURES
Microdilution-broth based antifungal susceptibility testing (AFST)
Predicting therapeutic outcomes as well as guiding the antifungal drug discovery process based on AFST of pathogenic fungi remains challenging. In a recent review, Sanguinetti and Posteraro presented an overview of standard AFST methods, focusing on their advantages and disadvantages, as well as of new promising technologies and newer-generation methods (e.g. whole genome sequencing) that can predict resistance [13].
–
The Clinical and Laboratory Standards Institute (CLSI; formerly the National Committee for Clinical Laboratory Standards [NCCLS]) and The European Committee on Antimicrobial Susceptibility Testing (EUCAST) have developed reproducible methods for testing the activity of antifungal agents against yeasts (the CLSI M27, M44, M60 and the EUCAST E.Def 7.3 documents) and filamentous fungi (molds; the CLSI M38, M44, M51, M61 and EUCAST E.Def 9.3 documents) [17][18][19][20][21][22][23]. These reference AFST methods, or their commercial counterparts such as Sensititre YeastOne (SYO, Thermo Fisher Scientific, MA, USA) rely on measuring growth of a defined fungal inoculum in a specific growth broth in the presence of different concentrations of the antifungal drug and allow the determination of the MIC (the minimum inhibitory concentration) of the drug resulting in complete or prominent growth inhibition. Clinical breakpoints have been determined by CLSI for anidulafungin, caspofungin, micafungin, fluconazole, and voriconazole against the prevalent Candida spp. whereas the EUCAST has set breakpoints for amphotericin B, anidulafungin, micafungin, fluconazole, itraconazole and voriconazole against the common Candida spp. and for amphotericin B, itraconazole, isavuconazole, posaconazole and voriconazole against the most common Aspergillus species [23][24]. These AFST methods deliberately minimize measurement of tolerance or trailing growth (see further) because it is highly variable under different culture conditions. For drug-organism combinations for which clinical breakpoints are not available, epidemiological cutoff values are suggested based on normal ranges of susceptibility of wild-type populations [16]. These should encompass the range of normal strain to strain variation within a species but exclude those organisms with known resistance mechanisms. Note that in some cases, published proposed epidemiologic cut-off values (see below) do not seem realistic. For example, for C. auris and fluconazole, they are so high that they fail their assignment as resistant. This has been recently addressed by the suggestion of more realistic, lower fluconazole breakpoints [25]. The occurrence of resistance is often associated with a genetic difference between a susceptible and resistant isolate. Resistance may be also the result of transient and reversible adaptation [26][27].
–
Commonly used MIC end-point terminologies are: The minimum inhibitory concentration (MIC) is the lowest concentration of an antimicrobial agent that prevents or inhibits the visible growth of fungal cells, as established by a standardized endpoint. Standardized MIC endpoints include partial inhibition of growth relative to the growth observed in the control (>50% inhibition of growth as determined visually [CLSI] or spectrophotometrically [EUCAST] for azoles and echinocandins), complete inhibition (100%) of visual growth (CLSI) or >90% by spectrophotometer (EUCAST) as applied to amphotericin B [28]. NOTE that MIC50, MIC90 and MIC100 are NOT appropriate in this context. The MIC50 is the concentration of an antimicrobial agent at which 50 percent of the strains of an organism tested are inhibited. The MIC90 is the concentration of an antimicrobial agent at which 90 percent of the strains of an organism tested are inhibited. The term minimum effective concentration (MEC) is used to describe the effect of echinocandin agents on filamentous fungi and is defined as the lowest concentration that results in conspicuously aberrant growth as assessed microscopically. Aberrant growth of hyphae is defined as small, round, compact microcolonies, often with swollen ends to the hyphae, compared with the matt of hyphal growth in the control well that does not contain an antifungal agent.
–
Interpretation of the efficacy of a given antifungal drug, is determined by the use of clinical breakpoints (CBPs). The CLSI uses the term ‘breakpoint’ as ‘clinical breakpoint’ is redundant in light of the fact that breakpoints are only applicable under clinical conditions. Thus the CBPs for in vitro susceptibility testing are used to indicate those isolates that are likely to respond to treatment with a given antimicrobial agent administered using the approved dosing regimen for that agent [28]. CLSI and EUCAST have established species-specific CBPs for some of the systemically active antifungal agents [CLSI [18], M60 Ed1; EUCAST E.Def 7.3 and E.Def 9.3]. The CBPs also provide information on the sensitivity of the CLSI/EUCAST methods to detect emerging resistance associated with acquired or mutational resistance mechanisms. The CBPs sort isolates into interpretive categories of susceptible (S), susceptible dose dependent (SDD; i.e. susceptibility is dose-dependent), intermediate (I), and resistant (R). The SDD category encompasses those organisms with MICs in a range that may respond to systemic therapy providing the drug levels in the blood are sufficiently high, especially relevant for fluconazole and voriconazole. For fluconazole and Candida glabrata, SDD is representing MICs < 32 µg/ml.. CBPs are established by taking into account microbiological (MICs and ECVs), clinical, molecular mechanisms of resistance, biochemical, pharmacokinetic and pharmacodynamic (PK/PD) data and provide the best cutoff value to predict clinical outcome for the treatment of a specific organism and antifungal agent [28].
–
Epidemiologic cutoff values (ECV/ECOFF) have been established to aid in the interpretation of MIC results when the lack of clinical data precludes the establishment of CBPs (CLSI, M57 and M59 [29] [30]; EUCAST). As mentioned above, they are also the first for proposing BPs. It has been suggested that some CBPs may not detect known mutational resistance in different species. Because of that, an extensive effort has been undertaken to establish ECVs or the MICs/MECs that separate wild-type (WT) from non-WT strains; the latter are more likely to harbor acquired and mutational resistance mechanisms. ECVs as BP are species-and-method-specific [30] and are also useful for tracking the emergence of strains with decreased susceptibility to a given agent and therefore less likely to respond to therapy [30]; ECVs can be used for the same purpose in surveillance studies [16]. Epidemiologic cutoff values can also be used to identify isolates that are less likely to respond to therapy when clinical CBPs cannot be established because of the rarity of infection with unusual species of fungi. However, some PK/PD must be known about the bug/drug combination in order to determine the efficacy of a given ECV/ECOFF and account for intrinsic resistance.
–
When testing the antifungal activity of a novel antifungal agent, relative to standard antimycotics, we propose to use the abbreviation ‘IC50 or IC90’, defined as the minimum inhibitory concentration of the agent or standard antimycotic that inhibits growth of the fungus (or similar readout like metabolic activity, see further) by 50% or 90%, respectively. In this way, there will not be a mix up with MIC abbreviations, which can only be used in the context of standardized AFST assays and endpoints.
–
One of the major drawbacks with AFST methodologies described above is that they are time-consuming and/or have long turn-around-times. Another concern is the general subjectivity involved in reading MIC end-points and the inter-laboratory variability of MIC values, especially for methods involving visual endpoint reading and for specific antifungal drugs such as caspofungin. EUCAST testing is advantageous for yeast as an objective spectrophotometric endpoint reading is performed [23] and image analysis of disk diffusion assays [31] or measurement of optical density (OD) in broth microdilution assays can provide a normalized quantitative measure of the degree of growth inhibition in the presence of a drug relative to growth in the absence of drug. EUCAST recommends reading the MICs based on OD measurements using a wavelength of 530 nm, although other wavelengths can be used e.g. 405 nm or 450 nm.
–
Promising alternatives to the classical phenotypic AFST are phenotype-centered (or semi-molecular) approaches that combine a culture step with molecular analysis (i.e. by real-time PCR or matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS)) [13]. However, in contrast to phenotypic methods, an important caveat of using PCR/sequencing is that it is suitable to detect resistance but not susceptibility as it can only detect resistance mechanisms that are already recognized. While the MALDI-TOF MS step of the analysis provides rapid analysis, the requirement for pre-assay cultured growth of the pathogen limits the ability to improve turn-around-times for more rapid diagnoses.
–
Fungicidal activity testing
Fungistatic drugs are defined most stringently as those that inhibit growth, whereas fungicidal drugs essentially kill all (>99.9%) cells in a fungal population. Typically, fungal pathogens are efficiently eliminated by the immune system in an immunocompetent host whereas immunosuppressed individuals are highly predisposed to fungal infections. Therefore, fungicidal drugs are invaluable for this vulnerable patient population for eliminating fungal pathogens [32][33]. Fungicidal drugs have an advantage over fungistatic drugs in that drug resistance is not common; however, the distinction between fungicidal and fungistatic activities could be problematic [34][35][36]. For example, although caspofungin is fungicidal for most yeast species, in molds it disrupts the hyphal tips but the surviving mycelium can continue to grow in the presence of the drug [37].
–
The minimum fungicidal/lethal concentration (MFC/MLC) has been defined as the minimum concentration resulting in a 99.9% reduction of fungal cell counts after a fixed time of incubation. Although arbitrary, the use of 99.9% (or 3-log-unit decrease) killing of the initial inoculum is the most stringent in vitro criterion for determining fungicidal activity [33]. In 2003, Canton and colleagues proposed a minor modification of the M27-A2 MIC procedure to allow MFC determinations based on the MIC test setup [38]. A smaller inoculum of 104 CFU/ml was used and the entire contents of each clear well in the MIC test were spotted onto two 90 by 15 mm Sabouraud dextrose agar plates (100 µl/plate), thereby allowing the fluid to soak into the agar. After the plate was dry, it was streaked uniformly to separate cells and remove them from the drug source. This method has been used in various studies for determination of MFC [39].
–
Similarly, time-kill assays can also be informative [39][40][41][42]. For Candida spp., these are typically carried out in 10 mL RPMI 1640 inoculated with 1-5 x 103 CFU/mL and concentrations of 32-, 16-, 8-, 4-, 2-, 1- and 0.5x the MICs. At predetermined time points (0-, 6-, 12-, 24-, 36- and 48 h), aliquots of 100 µl are removed from each control (drug-free) and test solution tube and then serially diluted in sterile water. A volume of 100 µL from serially diluted aliquots is placed on SDA plates to determine the number of CFU/mL after incubation at 35°C for 24 h. Using this method, Scorneaux and colleagues determined the time to reach 50%, 90%, and 99.9% reduction in the number of CFUs from the starting inoculum [42]. Net change in the number of CFU per milliliter was used to determine 50% and 90% effective concentrations and maximum effect (EC50, EC90, and Emax, respectively). Caspofungin was used as a fungicidal reference. Final DMSO concentrations were typically ≤1% (vol/vol) of the solution composition. Slight variations on this method use a more concentrated inoculum (105 CFU/mL) and/or smaller volume (5 mL) [43].
–
An alternative approach to perform time-kill studies using a BioScreen C MBR setup was implemented by Gil-Alonso and colleagues [44] where a final volume of 200 µL and an inoculum of 1×105-5×105 CFU/mL were used. At 0, 2, 4, 6, 8, 24, and 48 h, aliquots of 6 or 10 µL were removed from both the control and each test solution well, and were serially diluted in phosphate-buffered saline (PBS), and plated onto Sabouraud agar to determine the number of CFU per mL. For evaluating natural salt-sensitive antifungal peptides such as salivary histatins, defensins and lactoferrin, it is less clear what method is most suitable for assessing their activity, which is typically quenched in high salt media such as RPMI. In this case, kill studies are performed using yeast cells diluted in 10 mM pH 7.4 sodium phosphate buffer (NaPB) to 1 X 106 CFU/mL; then mixed with different concentrations of biological peptides at 30°C for 30 and 60 min with gentle shaking. Following incubation, samples are diluted in 10 mM NaPB, then aliquots of 500 cells are spread onto YPD agar plates and incubated for 36-48 h, until colonies can be visualized [45][46].
–
For filamentous fungi, Espinel-Ingroff and colleagues [47] tested several conditions for optimum determination of MFC, which were subsequently adopted by other research groups [48]. In these studies, the CLSI M38-A broth microdilution methods were used for MIC determination; following a 48-h incubation, 20 µL from each well without visual growth were plated on agar (Sabouraud dextrose agar) plates and MFCs were defined as the lowest drug concentration that yielded fewer than three colonies (approximately 99 to 99.5% killing).
–
Thus far, all the methods described for determination of fungicidal activity are dependent on the enumeration of replication-competent (i.e. culturable) cells following exposure to an antifungal agent. The inclusion of dyes validated for specific detection of either vitality or mortality in the conventional AFST MIC setups can be highly relevant for assessing the fungicidal nature of an antifungal in a direct way [33][49][50]. For example, propidium iodide (PI) is a fluorescent dye that can cross only permeable membranes and fluoresces upon interaction with DNA in dead, permeable cells. Hence, PI-positivity is generally regarded as a measure of cell death. However, it is noteworthy that cells in which apoptosis is induced by antifungal compounds, although dead, are typically PI-negative [51]. Of further note here is the fact that PI-positivity is not always indicating cell death as this can sometimes be restored to PI-negativity under certain conditions. In addition to fluorescent staining, tetrazolium compounds MTT (2H-Tetrazolium, 2-(4,5-dimethyl-2-thiazolyl)-3,5-diphenyl-, bromide) or XTT (2H-Tetrazolium, 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-hydroxide) are also commonly used to assess cell viability (with these dyes redox potential is measured as a proxy of viability) [52]. Similarly, actively respiring cells convert the water-soluble XTT to a water-soluble, orange colored formazan product. However, unlike MTT, XTT does not require solubilization prior to quantitation, thereby reducing the assay time in many viability assay protocols. Similarly to the MTT-to-formazan reduction, the addition of an oxidation-reduction colorimetric indicator like Alamar blue (containing resazurin) can be used. This indicator changes from blue to the pink fluorescent resorufin in the presence of metabolically active growing cells [53]. A disadvantage of resazurin can be its low level of stability when incubated for 7 days or longer (required for certain dermatophytes). As discussed further below, these dyes are most often used to assess the effect of antifungal agents against biofilms.
–
Additional comments and notes with respect to MIC/MFC determinations
For studies assessing efficacy of antifungal therapy, resistance development during therapy and fungal epidemiology, it is necessary to determine clear and uniform MIC/MEC/MFC end-points according to the reference AFST methods indicated above. However, this is not necessarily the case for the identification of novel antifungal compounds during early preclinical drug discovery phases. For the latter, compliance and feasibility for high-throughput testing (either compound libraries or deletion mutant collections for investigating mode of action) are generally more important than exact end-point MIC/MEC/MFC, as long as the MIC/MFC end-points of relevant standard antimycotics are included and used for interpretation of relative activities. Deviations from reference AFST methods generally relate to different media/buffers; inclusion of serum might be most relevant when screening for fungicidal compounds that retain activity in the presence of serum; and inoculum size. The latter might need to be adjusted when using higher throughput setups based on for instance a Bioscreen apparatus.
–
One of the most puzzling effects that can confound MFC/MEC/MIC determination is the paradoxical effect or Eagle effect which has also been described with antibacterial agents. This effect is defined as the ability of the fungus to grow at high antifungal concentrations (above the MIC), but not at intermediate concentrations [43][54][55][56]. For example, a paradoxical effect for echinocandins against various C. albicans isolates and against A. fumigatus has been demonstrated. Paradoxical growth varies in terms of media, species, strain and type of echinocandin and is also of specific concern in the biofilm field (see further). Rueda and coworkers demonstrated that paradoxical growth of C. albicans is associated with multiple cell wall rearrangements and reduced virulence [57]. It is important to note however, that observed in vitro paradoxical growth does not necessarily indicate lack of response to the antifungal drug in vivo [58]. Therefore, the clinical implications of fungal adaptation against antifungal drugs, which might be linked to reduced virulence, still remain to be elucidated. In addition, MIC/MEC deviations can be induced by the presence of active volatile compounds in neighboring wells of a microtiter plate. By screening a large collection of essential oils it was shown that ‘paradoxical like phenotypes’ were caused by small volatile molecules released from adjacent wells. This observation led to the development of a quantitative method to evaluate the activity of volatile molecules in the vicinity [59].
–
Another unclear issue has been the description of drug responses as tolerance or trailing growth, which has often been measured using modifications of MIC assays and is particularly relevant for azole and echinocandin (both Candida spp. and filamentous fungi) activity against Candida spp [60][61][62][63]. Tolerance may be perceived as a subpopulation effect that is due to slow growth of a subpopulation of cells in a manner that is generally drug-concentration independent within the range of concentrations studied [64]. Of note, standard MIC values are determined following at 24h growth period; however, tolerance may become evident after 48h of growth in the presence of the drug. Tolerance can also be detected by agar disk diffusion assays [31][64]. Recently, additional definitions have been proposed in this context, namely fraction of growth (FoG) inside the zone of inhibition and the Supra-MIC growth (SMG) in drug concentrations above the MIC. The FoG and SMG measurements correlate with one another and are clearly distinct from measurements of the MIC or the radius of the zone of inhibition [31][64].
METHODS FOR ANTIFUNGAL SUSCEPTIBILITY TESTING OF BIOFILMS
Microbial biofilms consist of dense layers of microorganisms surrounded by an extracellular polymer matrix, thereby protecting the microbes from the action of antimicrobial agents. In addition, dormant persister cells have been proposed to make up a small proportion of some microbial cultures (either a biofilm or free-living culture) that can withstand the action of high doses of most antimicrobial agents and may facilitate the recurrence of microbial infections after treatment ceases [65][66]. In medical mycology, the appearance of persister cells is often referred to as ‘heteroresistance’, as the cells are not resistant in the classical sense. Rather, they transiently acquire the ability to survive and grow at normal growth rate under conditions that inhibit growth of the majority of the isogenic susceptible population [67][68][69][70]. Heteroresistance is defined as a small proportion of the population (usually <1%) that is able to grow in the presence of the drug. This is different from tolerance or trailing growth, where many cells in the population (>1%) are able to grow, albeit slowly, in the presence of the drug.
–
The majority of C. albicans infections are associated with biofilm formation [71][72][73][74][75]. A. fumigatus is the most important airborne human fungal pathogenic mold and the number of chronic A. fumigatus infections is constantly increasing in patients suffering from respiratory tract diseases. Until recently, most studies undertaken to understand Aspergillus physiology and virulence were performed under free-living conditions. However, in all Aspergillus infections, A. fumigatus grows as a colony characterized by multicellular and multilayered hyphae that are in some cases embedded in an extracellular matrix (ECM). This type of growth is generally consistent with the definition of a biofilm [76]. Therefore, it is becoming increasingly important to also assess the activity of current antimycotics and novel antifungal compounds against biofilms.
–
Several methods for fungal susceptibility testing under biofilm conditions for determination of sessile MICs (SMICs) have been developed which in principle are based on methods for planktonic culture testing [13][71][76][77]. The most commonly used method is based on a static model using 96-well microtiter plates [78][79], thereby quantifying microbial biomass or metabolic activity, using compounds such as crystal violet (CV), 2,3-bis (2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-tetra- zolium hydroxide reduction (XTT), the more reliable fluorescein diacetate (DFA), or resazurin [80]. Though, caution should be exercised in the particular assay employed, as each assay has its pros and cons depending on the question being addressed, e.g. XTT should only be used to compare the effect of an active within a specific strain and not to compare different strains [81]. Propidium iodide (PI), described above, is also used to assess the degree of killing of biofilm cells. Recently, a biofilm model for A. fumigatus was reported, in which conidia and hyphae are trapped in an agar layer with measurement of metabolic activity by the XTT assay [82]. A newer high-throughput approach grows biofilms with shaking in 96-well and 384-well plates in the presence of the compound of interest, and uses optical density at 600nm (although other wavelengths can be used) as a readout of the biofilm remaining during or after exposure to the compound [83]. As indicated by the authors, the optical density of the biofilm correlates with the number of viable cells in the biofilm, and thus is an accurate read-out of biofilm formation in the presence of the compound of interest. This method returns consistent results with less labor and can be performed in high-throughput. A more labor-intensive approach for analysis of C. albicans biofilms involves determining the number of CFUs recovered from treated biofilms, which involves harvesting the biofilm by scraping, homogenizing and plating homogentaes on agar media for colony enumeration. Homogenization can be done by vortexing (potentially in the presence of 1% Triton) or sonication. However, these procedures must ensure that all biofilm cells are harvested and individually separated without affecting the viability of the biofilm cells, three conditions that can be difficult to achieve. Another limitation of cultivation-based assays lies in the fact that biofilms contain hyphae which might bias CFU counts. Ideally, measurements should rely on nucleus counting which can be performed using qPCR. In theory, it is recommended to use at least two assays that rely on different methods or dyes as a readout.
–
While static biofilm models in 96-well plates are used most frequently, continuous flow models that better mimic in vivo situations can be used to assess C. albicans biofilm formation and inhibition [84][85]. Unlike static biofilms, C. albicans cells under constant laminar flow undergo continuous detachment and seeding that may be more representative of the development of in vivo biofilms [86]. A particularly relevant example is the study of oral biofilms, which form in the presence of salivary flow. Most notably, it was shown that biofilm cell detachment rates are an important predictor of ultimate biofilm mass under flow [86]. However, whether these flow models are indeed better will depend on the pathogenesis of the infection under study.
–
Microscale technologies, such as microfluidics provide a more revolutionary approach to study biofilm formation in dynamic environments. By enabling control and manipulation of physical and chemical conditions, these technologies can better mimic microbial habitats in terms of fluid flow and nutrient sources [87]. Gulati and coworkers recently described a protocol to study biofilm formation in real-time using an automated microfluidic device under laminar flow conditions [88]. This protocol enabled the observation of biofilms in real-time, using customizable conditions that mimic those of the host, e.g., conditions encountered in vascular catheters. This protocol can be used to assess the biofilm defects of genetic mutants as well as the inhibitory effects of antimicrobial agents on biofilm development in real-time. Most recently a novel technique consisting of nano-scale culture of microbial biofilms on a microarray platform was developed whereby thousands of microbial biofilms, each with a volume of approximately 30-50 nanolitres are simultaneously formed on a standard microscope slide. Despite a 2000-fold miniaturization compared to microtiter plates, the resulting nanobiofilms display similar structural and phenotypic properties. This technique platform can significantly speed up biofilm susceptibility testing and allows for true high-throughput screening in search for new anti-biofilm drugs [89][90][91].
–
In addition to the SMIC, the BIC-2 (BIC50), is the minimum compound concentration resulting in a 2-fold inhibition of biofilm formation. Similarly, the BEC-2 (BEC50) is the minimum compound concentration resulting in a 2-fold eradication of mature biofilms [92][93]. In practice, to assess biofilm inhibition, compounds are included during the biofilm growth phase. To assess biofilm eradication however, biofilms are grown for 24h after which the compounds are added and biofilms are additionally incubated for another 24h in the presence of the compounds. In that respect, BEC determination is most relevant for compounds that can kill biofilm cells, whereas BIC determination is relevant for compounds that might only inhibit adhesion without impacting viability of biofilm cells. Hence, biofilm inhibiting compounds can be very relevant for the design of antibiofilm material coatings (see further), whereas biofilm eradicating compounds can be used to design curative antibiofilm therapy. With regard to terminology, SMIC20/50/80 can also be used (see before): SMIC80 has been defined previously as an 80% reduction in the metabolic activity of the biofilm treated with the antifungal compared to that of untreated biofilms [60]. In general, SMICs can be up to 1000-fold higher than the corresponding MICs for a particular antifungal agent [94]. Among the different mechanisms that may be responsible for this intrinsic tolerance of Candida species biofilms are: the high density of cells within the biofilm; nutrient limitation within the biofilm; effects of the biofilm matrix; antifungal resistance gene expression; and the increase of sterols in biofilm cell membranes [95][96].
–
Although it is clear that a wide range of (standardized) techniques is available to determine the activity of compounds against biofilms, there is currently little evidence that implementing these biofilm-based assays in the clinical microbiology laboratory lead to better treatment outcomes [97].
–
Synergistic antifungal/antibiofilm drug combination testing
In addition to screening for novel antifungal compounds, combination therapy is considered a potential alternative strategy for treating invasive fungal infections [98][99]. In general, the main objective of combination therapy is to achieve a synergistic interaction between two compounds, thereby increasing their activity and reducing potential toxic effects of each compound. Apart from combining two antifungal (or antibiofilm) agents that are characterized by different mode of actions that can synergize each other, another option is to combine an antimycotic with a non-antifungal potentiator. An increasing number of studies document the synergistic action of such antifungal-potentiator combinations, with the potentiators being (lasso)peptides like antifungal tyrocidines, humidimycin, or plant defensins, or repurposed compounds like toremiphene or artesunate [92][100][101][102][103][104][105][106]. Repurposing of known drugs, i.e. finding novel therapeutic indications for existing drugs, is favorable from an economic perspective, as these molecules are often FDA-approved and have a known (and often safe) toxicity profile and dosing regimens are known. Furthermore, the cost of performing new clinical trials with existing drugs with possibly reformulating the drug are likely to be far lower than the costs of developing a new drug.
–
An interaction between two compounds is defined as synergistic when the combined effect of the two compounds is greater than the sum of their separate effects at the same doses. Synergistic efficacy can be quantified in vitro using checkerboard assays, where two-fold dilution series of one compound are combined with two-fold dilution series of the other compound and scored for the read-out of interest (e.g. growth inhibition, killing, biofilm inhibition, biofilm eradication). The fractional inhibitory concentration index (FICI) is determined as d1/(D1)p + d2/(D2)p, where d1 and d2 are the doses of compounds 1 and 2 in combination to result in, for example, 50% inhibition of biofilm formation and (D1)p and (D2)p are the doses required for the two respective compounds alone to produce the same effect. An interaction is scored as indifferent/additive if 0.5<FICI<4; as antagonistic if FICI >4 or synergistic if FICI <0.5 [107].
–
Although the FICI is most frequently used to define or describe drug interactions, it has some important disadvantages when used for drugs against filamentous fungi. This includes observer bias in the determination of the MIC and lack of agreement on the endpoints (MIC-0, MIC-1, or MIC-2 [≥95, ≥75, and ≥50% growth inhibition, respectively]) when studying drug combinations [108]. Moreover, when one compound strongly potentiates the other but the reverse is not the case, the FICI value will not reach values below 0.5. Synergy then is not concluded, whereas there certainly can be a very relevant reduction in concentration of major antifungal agents, with concomitant reduction of toxic effects in (prolonged) treatment.
METHODS FOR TESTING OF MATERIALS AND COATINGS THAT RESIST FUNGAL BIOFILM FORMATION
Fungal adhesion and subsequent biofilm formation on biomedical implants and devices are a major cause of biofilm-associated infections. Because treatment of such infections is very difficult (see previous section), emphasis has shifted to the prevention of these infections by the design of antimicrobial coatings for biomedical devices. In general, to prevent microbial biofilm formation, coatings rely on either of three principles: they can act via release of antimicrobial agents, via coating of antimicrobial agents (resulting in contact-cidal activity) or via coating of anti-adhesive agents that are not antimicrobial. In a recent review by Sjollema and colleagues [14], 15 methods or groups of methods to assess in vitro performance of these three types of antimicrobial coatings were discussed. To evaluate the efficacy of one of the three antimicrobial designs independently, no single method could be determined to be “one for all” with all having different merits for different antimicrobial designs.
–
Many antimicrobial designs in clinical practice and described in literature are based on the slow or fast release of antimicrobials [109]. By far the most applied methods to evaluate the effect of antimicrobial release on microbial growth inhibition are “agar zone of inhibition“ methods [110][111], very similar to disk diffusion susceptibility tests as described by the CLSI standard for yeasts (CLSI M44-A). In these methods, a plate with nutrient agar media is inoculated with microorganisms and a test sample is subsequently placed with the antimicrobial side on the agar. Following an incubation period during which released antimicrobials from the test sample diffuse into the agar, a zone of growth inhibition is formed the diameter of which is indicative of the level of susceptibility of the microorganisms to the antimicrobials and the amount released.
–
Although this method is effective, simple to execute and available in most microbiology labs, it does not quantify the antimicrobial efficacy [112]. For quantification of efficacy of surface designs based on release of antimicrobials, various assays are described which are mainly categorized as “suspension methods”. In these assays, a known inoculum of microorganisms, suspended in a nutrient medium, is exposed to a test sample and incubated for a set period of time (usually 1 to 2 days, but for moulds a longer period) [113]. Following incubation a sample of the suspension is taken and the number of surviving microorganisms is enumerated (for bacteria and yeasts often by CFU, for fungi quantitative colorimetric XTT assays are more appropriate [114]).
–
A variation of this suspension method is recommended in situations where a clinical scenario is characterized by a very small volume/area ratio, such as in the narrow extraluminal space between a urinary catheter and the urethral epithelium, where released antimicrobials accumulate rapidly. Typically, in “high area to volume tests” a small volume of a microbial suspension is incubated, sandwiched between a thin cover sheet and the test sample, therewith stimulating intimate contact between the microorganisms and the sample [115]. Typical examples are the JIS-Z 2801 [116] and “all in one” plating systems (e.g. for yeasts and fungi Petrifilm® Yeast and Mold all-in-one-plating systems, 3M, St. Paul, MN, USA) [117][118]. Because the intimate contact between microorganisms and sample is established in these type of “high area to volume tests” they are often applied in evaluating contact killing designs [119].
–
Intimate contact is also established in “adhesion based assays” where microbial cells suspended in a low nutrient suspension are allowed to settle. Non-adhering organisms are subsequently removed by washing and adhering cells are counted microscopically or cultured following sonication [120]. Flow systems, a special adhesion-based assay in which microorganisms are exposed to a sample surface from a flowing suspension, mimic flow in clinical environments such as in the intraluminal area of a urinary catheter or the extraluminal area in intravenous catheters [121]. Another advantage of flow systems is that passage of samples by liquid-air interfaces and sonication applied as critical steps before assessment, can be circumvented in case of the application of real time microscopy since non-adhering microorganisms are continuously flushed away [118][122]. Adhesion-based methods are preferred in the evaluation of non-adhesive surface designs.
–
In order to study the efficacy of antimicrobial designs in preventing biofilm formation, biofilm methods are applied [123][124]. These methods resemble the “adhesion based” methods in that the process begins with an adhesion step in a low nutrient environment and over an extended incubation period, adhering microorganisms form a biofilm. In order to prevent microbial growth in the suspension, prior to the incubation step, non-adherent cells are removed by washing. The biofilm methods can be adapted to evaluate all types of antimicrobial designs.
–
Although several methods have been applied in research laboratories and industry, minimal guidance is provided on how to discriminate release, contact killing and non-adhesive systems. For instance, in the development of novel surface microbicidal coatings, it is important to accurately assess whether antimicrobial activity arises from direct contact or from inadvertently released compounds or components. Such differentiation may be key for successful translation to a product, as medical devices with non-releasing surface coatings are considered “pure” medical devices while release-systems are considered to be so-called combination products. Combination products entail a different regulatory pathway, i.e. the whole entire pharmaceutical activity of the released therapeutic product has to be considered requiring a battery of in vitro testing and extensive toxicology and safety assessment [125]. Therefore, there is a need for simple industry standards that allow discrimination between the various antimicrobial designs and in particular incorporate a presently lacking standard test for adhesion under flow conditions that resemble the flow occurring in various clinical applications.
METHODS FOR MONITORING IN VIVO PERFORMANCE OF ANTIFUNGAL AND ANTIBIOFILM DRUGS
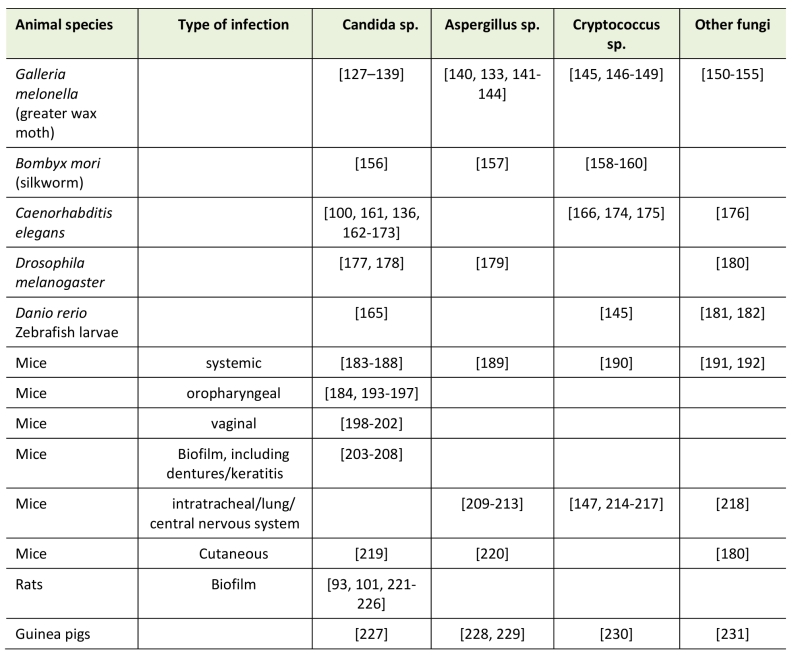
Compounds that perform well under in vitro testing conditions and show no or low toxicity should be validated under more physiologically relevant conditions. To this end, different animal model systems have been developed (see table 1) to evaluate efficacy of antimicrobial agents in vivo. However, in an effort to minimize the use of animals, three-dimensional organoid (ex vivo) tissue systems have been developed to screen for new drug candidates. This in vivo or ex vivo evaluation of candidate molecules is essential as many compounds that are very potent in vitro, fail the in vivo test [126]. Moreover, such testing is required in view of toxicity assessment and progression to clinical application.
–
Human three-dimensional organoid tissue culture approaches or ex vivo human tissue, that both can be combined with specific immune cells have been developed, mainly in the cancer field [232]. Recently a method was developed to mimic the large intestine where colonic organoids are generated from differentiated human embryonic stem cells or induced pluripotent stem cells [233][234]. This and similar approaches will likely be used in the future for disease modeling and drug discovery [234]. To study C. albicans infections, a human skin model was developed which can be used to test the effect of novel antibiofilm compounds, mainly for their effect on inhibition of C. albicans adhesion [235]. Similarly, an organotypic model of the human bronchiole was developed for testing host pathogen and three-way host, fungal and bacterial pathogen communication and immune response [236]. This model can also be used to test the effect of antimicrobial compounds during host-pathogen interaction.
–
The most relevant models however, remain animal model systems as these allow for the study of pharmacodynamics and pharmacokinetics of novel compounds (see table 1). In general vertebrate model systems are preferable as they are phylogenetically closer to humans; however, several lower, non-vertebrate, models have been optimized for virulence evaluation as well as for drug screening [15][237]. Among these model systems, the most used are Caenorhabditis elegans [161][238], Drosophilla melanogaster [177][179][239], zebrafish larvae (Danio rerio) [145][240], the silk worm Bombyx mori [158][157], the heat tolerant two-spotted cricket [241] and the larvae of the greater wax moth Galleria mellonella [140][242][243]. The main advantage of the latter two is that these organisms can be maintained and grown at 35 °C to 37 °C, approximately the same as the human body temperature.
–
Developing a relevant model system requires considerable effort and time, and some examples of how to set up Aspergillosis model systems for drug discovery are described by Paulussen et al. [244] and Lewis and Verweij [245]. When performing animal experiments to test the efficacy of drugs it is important to clearly define the endpoint in terms of organs or tissues positive for fungal presence at a particular time point during / after initiation of treatment, since the niche in the host might influence the outcome. A typical example is the pH dependent virulence of C. albicans, where a strain may not be virulent systemically but is virulent vaginally or vice versa [246][247][248]. Recently, it was shown that fitness cost, often associated with drug resistance, is also niche-dependent as fluconazole-resistant isolates were outcompeted in a gastrointestinal colonization model and not in a systemic infection model system [249].
–
Vertebrate model systems in use for testing novel antifungal drugs, are primarily based on those developed to investigate virulence mechanisms by fungal pathogens [250]. These model systems can be classified as methods to test therapeutic efficacy against superficial (skin, nails), mucosal (oral, vaginal), gastrointestinal, lung or systemic infections. In order to mimic the population at risk for certain types of fungal infections, in many cases animals are rendered immunosuppressed, which may influence the efficacy of a specific antifungal or compound used. Sex of the animals and route of administration of the pathogens will also determine the type and outcome of the infection. For opportunistic pathogens, such as Candida spp., various infection sites can be used that may either result in a systemic infection (intravenous, intraperitoneal, infections) or in local infections (oral cavity, gut, vagina or skin infections) [reviewed in [250]]. In addition to testing the effect of antifungal drugs, animal model systems are also used to investigate the pharmacokinetic, tissue distribution and stability of antimicrobial drugs [251][252].
–
Superficial infection models
Different animal model systems have also been developed to test the efficacy of antifungals against dermatophytes and other fungi, such as C. albicans that can cause skin infections [253][254][255]. The guinea pig is the standard model system for these type of skin, hair and nail infections but mouse model systems have also developed such as for C. albicans [219][256] and A. fumigatus [220][257].
–
Experimental induction of dermatophytosis in laboratory animals has been performed for decades and early publications date from the 1960s [258]. Guinea pigs have always been one of the target animals as they tend to be predisposed for dermal fungal infections and consequently, artificial infection is easy and does not require any kind of preconditioning or immunosuppressant treatment [259]. To establish a dermatophyte infection in guinea pigs, different methods can be employed. When the model was first being used and described, it was believed that occlusion of the inoculated area was essential to induce an experimental infection [260]. In the more recent publications, however, occlusion is only rarely applied and was even demonstrated not to offer any advantages on the establishment of a skin infection [253]. General agreement exists on the necessity of prior trauma of the skin, before applying the inoculum. Skin damage can be achieved by using a manual razor or tape stripping, or scarification with a pumice stone, sandpaper or other methods. The most frequently used fungal species to induce experimental dermatophytosis are Microsporum canis and Trichophyton mentagrophytes, both zoophilic species but also common causative agents of human infections [259]. The inoculum size usually varies between 1×105 to 1×107 CFU/ml [253][259]. In contrast with dermatophyte infections in humans, animals show spontaneous clearance of this disease with formation of a partial immunity against reinfection [261]. Methods to evaluate antifungal efficacy involve microscopic or culture examination of skin scrapings, skin biopsies, scales and/or hair samples, and histopathology of skin biopsies, while other groups use a scoring system [262][263].
–
Onychomycoses are fungal nail infections responsible for 50% of all nail dystrophies and mostly caused by dermatophytes, of which Trichophyton rubrum and T. mentagrophytes are the most important ones. Most of the studies on the biological activity of topical antifungals are based on in vitro models, since the existing rabbit [264] and guinea-pig [265] models are extremely time-consuming, labour-intensive and expensive.
–
Mucosal infection models
Vulvovaginal candidiasis (VVC) is one of the models used to study mucosal infection with Candida spp., especially C. albicans. Vaginal candidiasis has been studied in mice, rats, guinea pigs and rabbits with mice and rats appearing the more sensitive species and therefore most commonly used for this type of infection. More so, the rat model offers more accurate data compared to the mouse model and is therefore the better model to study antifungal efficacy. However, like most mucosal models of candidiasis, the establishment of VVC requires predisposition of the animal, which is usually obtained by ovariectomy and hormone injections to maintain permanent oestrus, although immunosuppression or antibiotic treatment have also been employed [266]. For infection, a cell suspension is directly introduced into the vagina, and disease development is assessed based on microbial recovery (CFUs) from vaginal swabs. However, histopathology and immunological analysis can also be performed with this model [231][267][268].
–
For studying oral candidiasis, infections are induced by placing a swab impregnated with fungal cells for a specific period of time under the tongue or rodents, and antimicrobial efficacy is assessed based on the level of microbial recovery and histopathology of infected tissue [269]. Recently a model was described to induce concurrent oral and vaginal mucosal Candida infections [270].
–
Lung infection models
For airborne infections (Cryptococcus spp., Aspergillus spp., dimorphic fungi) the model of choice is the mouse lung infection model where on average 104 fungal cells are administered through inhalation, intranasal or intratracheal administration [271]. Antifungal treatment is generally initiated 24-48 hours post-infection, although starting at earlier time points (after several hours or 1 day) is also reported. The current standard to treat such infections is amphotericin B (AmB) and this can be used as a control (0.5mg/kg/day) [272]. This resembles the current treatment of such infections in humans where a combination therapy consisting of AmB and 5-fluoro cytosine is used to treat Cryptococcus central nervous system infection [273] and voriconazole is used to treat aspergillosis [274]. Apart from pulmonary infections, Aspergillus sp. can cause a wide variety of infections and for several of them, relevant animal model systems have been developed. Some examples include the development of a sinusitis infection model [209], a rat model to study asthma [275] and a guinea pig endophthalmitis model [228]. In addition to Cryptococcus and Aspergillus, model systems for other environmental fungi have also been developed such as a fusariosis model system [276], a sporotrichosis model system [277], a mucorales model system [278][279] and a model system for Scedosporium [280] (see Table 1). Efficacy of treatment of these fungal infections is often performed by non-culture based methods, such as qPCR to determine organ fungal burden [281].
–
Gastrointestinal infection models
Gastrointestinal infections are induced by oral gavage of fungal cells. To establish a colonization, first the endogenous microbiota is partially cleared by administration of antibiotics, followed by a single oral gavage with C. albicans and stool samples are analyzed to determine colonization. Following euthanization segments of the gastrointestinal tract can be harvested and analyzed for CFUs (or BLI, see further) [282].
–
Gastrointestinal colonization model systems can also be used to induce a systemic infection which is reflective of one of the natural routes of a systemic infection in humans [283]. Recently a model was described where an oral infection was initially established in mice administered tetracycline and prednisolone, which was followed by an infection of the small intestine and subsequent systemic infection [284]. Specific diet-based mouse models for systemic infection via the gastrointestinal tract have been developed [285]. Treatment of the animals with either fluconazole or echinocandins resulted in a significantly reduced organ microbial burden, and therefore this model would be suitable to study the efficacy of drugs in inhibiting of dissemination from the gastrointestinal tract. Interestingly, gastrointestinal tract infections have also been shown to lead to echinocandin resistance in C. glabrata when used in a mouse model of colonization and systemic dissemination [286]. Therefore, these models are also ideal to test novel antifungal compounds or combinations of compounds. C. albicans is normally not part of the microbiota of the gastrointestinal tract in mice and therefore, it is not an animal model that can be colonized by C. albicans under natural conditions. However, the group of Lois Hoyer showed that piglets are naturally colonized by C. albicans and successfully used this model for gastrointestinal infections, although therapeutic studies have not yet been performed in this model [287].
–
Systemic infections
Systemic infections with C. albicans are mostly induced using the mouse tail vein infection model system, where between 104 and 106 Candida cells are injected in the lateral tail vein [288][289]. Whereas for normal virulence assays the inbred lines BALB/c or C57bl/6 are mainly used, for antifungal drug screening, outbred lines such as ICR or CD-1 should be used [183][290].
–
Antifungal therapy can be administered prior to infection, on day of infection, or at later time points post-infection and depending on the compound, administration could be via the oral, intraperitoneal or intravenous route. It is important here to iterate that the inoculum dose is critical; for example, the difference between the most virulent and the least virulent strain of C. albicans can be compensated for by adding a 10 fold excess of the latter as the inoculum, in a systemic mouse model of infection [291]. Survival curves, animal weight and/or tissue burdens are determined as a measure for the efficacy of the drugs [289]. The tail vein model system has been used to test the efficacy of different antifungal drugs using different Candida ssp. as well as other fungal pathogens [292][293][294].
–
Pharmacokinetics
When discussing dosing in animal models, it is important to consider that elimination of fungal pathogens in animals and humans may vary greatly. Hence, comparing activities of two agents in an animal model using doses extrapolated from those used in humans based on weight may be misleading, as the antifungal exposure may not correspond to the one achieved in humans. This is an important caveat when comparing drugs to each other unless it is documented that the bioavailability is comparable. A case in point, comparing the activity of echinocandins in rodent model systems will not fully represent the activity in humans because the clearance and the volume of distribution is different between e.g. rats versus primates/humans. In fact, the clearance of all three echinocandins is approximately 6-fold higher in rats than in humans [295][296][297][298]; specifically, caspofungin and anidulafungin have a volume of distribution that is 2- to 3-fold higher, compared to that in humans, and in micafungin this is only slightly higher in rats compared to humans [297]. This difference is probably the reason why in rats higher concentrations of micafungin were required compared to caspofungin and anidulafungin to clear C. albicans biofilms in a subcutaneous biofilm model system [223]. Also, voriconazole is rapidly metabolized in mice and rats due to autoinduction of cytochrome P450 [299]. Thus, rodents require much higher doses of voriconazole than human to achieve similar drug exposure.
–
Kinetic experiments to monitor the efficacy of drug treatment over time require large numbers of animals when CFU counting is used to analyze the effect of the drugs or survival. To overcome this issue, various imaging approaches including fluorescence and bioluminescence have been developed as they allow longitudinal experiments in single animals. For this purpose, C. albicans and C. glabrata strains have been engineered to express luciferases in order to perform bioluminescence imaging [300]. Gaussia princeps and firefly luciferases have been codon-optimized for use in C. albicans and were used to monitor superficial infections such as vaginal and oropharyngeal infections [198][301][302], subcutaneous infections [303] and systemic infections [304]. Recently, a red-shifted firefly luciferase was optimized for bioluminescence imaging [184]. However, a limitation of the bioluminescence approach is that the presence of oxygen is required for the generation of light by luciferase. Thus, this approach will underestimate the fungal burden in established fungal lesions in which there is hypoxia. In addition to the bioluminescence approaches, fluorescence-based approaches were also developed such as the bronchoscopic fibered confocal fluorescence microscopy, which was used to assess pulmonary Aspergillus and Cryptococcus infections in live animals. This approach also allows longitudinal imaging of the same animal [305]. Computed tomography and magnetic resonance imaging was also used to assess invasive pulmonary aspergillosis over time in a single animal [306]. Correct diagnosis of fungal infections can be problematic, however recently imaging-based tools were developed to accurately diagnose infections such as fungal keratitis. Based on the structure of caspofungin a chemical probe exhibiting high affinity to Aspergillus and possibly also other fungal pathogens was developed which could serve as a basis for development of an imaging system [307].
–
Biofilm models
Biofilms are notoriously difficult to treat and eradicate and therefore, several biofilm model systems have been developed and used to test the efficacy of antibiofilm molecules [308][309]. The first model was a rabbit model of C. albicans biofilm-associated catheter infection using a catheter-lock system to determine the efficacy of antifungal treatment [310][311]. In the same year, Andes et al. developed a rat central venous catheter (CVC) model which was also used to test the efficacy of antifungals [72][193][203][312][313]. Although the CVC model has the advantage of closely mimicking the clinical situation in a patient, the disadvantage is that it is technically demanding and that only one catheter can be placed in an animal. To overcome this limitation a subcutaneous catheter rat model system was developed where in a single animal up to nine catheter fragments can be implanted. To develop a biofilm infection, catheter fragments are inoculated with C. albicans in vitro prior to implantation in the animals and biofilm is allowed to develop [314]. This model system was used to test the efficacy of mono or combination treatment with antibiofilm molecules [221][222][223] and was also adapted for use with C. glabrata [224]. Another rat model was developed to study Candida-associated denture stomatitis, a prevalent oral condition stemming from the ability of Candida to adhere to denture material and form biofilms with commensal oral bacteria [204][315][316].
–
Apart from traditional antifungal compounds, there is also a growing interest in the use of probiotic bacteria as agents against C. albicans. In a recent study, the antifungal activity of clinical isolates of Lactobacillus strains were tested against C. albicans biofilms in vitro and it was shown that certain species of lactobacilli had an effect on C. albicans morphogenesis and biofilm formation [317][318]. Using the G. mellonella model system the same group showed that these probiotic bacteria reduce filamentation by C. albicans and also stimulate the host innate immune system, thereby protecting the host against C. albicans infections [319].
–
In any of the above models, it is important to consider that fungal organisms often have metabolic and phenotypic distinctions in the context of biofilms as compared to non-biofilm settings. Parameters that may affect antifungal susceptibility in experimental biofilm models in vitro or in vivo include the effects of quorum sensing [320], impact of pH signaling in abscess biofilms [321], metallic stress as may be amplified in context of devices [322], influences of host factors including immune effectors [323][324] and other micro-environmental factors. Thus, a key goal of antifungal susceptibility modeling may be evolving to specialized assay conditions that most accurately correlate with or predict outcomes in specific clinical conditions, rather than universal testing systems.
METHODS FOR MONITORING IN VIVO PERFORMANCE OF ANTI-INFECTIVE MATERIALS
In vivo model systems have also been developed to test the efficacy of coated implant material against fungal and bacterial pathogens. A modification of the subcutaneous catheter biofilm model described above was used to test the efficacy of titanium discs coated with caspofungin, vancomycin or other small antimicrobial compounds. In this model, small (0.5 cm diameter) titanium discs that were coated with the antimicrobial compound through covalent attachment using a linker or only the linker as the negative control, were implanted subcutaneously in the back of immunosuppressed Wistar rats. The following day, animals were infected with either C. albicans or S. aureus cells in the area of the implanted discs and microbial adhesion on the discs were determined after 2 days (bacterial cells) or 4 days (fungal cells). Using this in vivo model, caspofungin- or vancomycin-coated discs harbored significantly lower numbers of adherent fungal and bacterial cells, respectively [325].
CONCLUSIONS AND FUTURE PERSPECITVES
The increasing impact of fungal infections on society is mainly due to the increasing population of patients at risk, as well as the rather limited armory of antifungal agents and resistance development. Therefore, there is a critical need for the identification and development of new antifungal agents, or antifungal combination therapies, particularly those that are also active against fungal biofilms and do not suffer from resistance development. To accomplish this, a set of standardized, simple guidelines describing the appropriate methods to assess the performance of novel antifungal and/or antibiofilm agents is warranted. Consistent with the recent emphasis on preventing infections, many efforts are focused on developing antibiofilm coatings for medical devices such as catheters and implants. Thus, standardized testing of such materials is of considerable importance.
–
To that end, in this review we aimed to provide a compilation of general methodologies and definitions for assessment of susceptibility of fungal species to antifungal agents, when grown as planktonic cultures or as biofilms focusing on the standardized AFST guidelines. However, more recent developments and technologies in susceptibility testing designed to better mimic in vivo conditions with respect to flow and nutrient conditions are also discussed. These methods are still in their infancy and new guidelines for their use will need to be established in the future.
–
There is a plethora of abbreviations to quantify an antifungal or antibiofilm effect (see box 1), and various abbreviations have been used in different contexts. However, we want to emphasize that the MIC can only be used in the context of standardized AFST assays and endpoints, mostly reference or methods comparable to those methods. When testing the antifungal activity of a novel antifungal agent, relative to standard antimycotics, we propose to use the abbreviation ‘IC50 or IC90’, defined as the minimum inhibitory concentration of the agent or standard antimycotic that inhibits growth of the fungus (or similar readout like metabolic activity) by 50% or 90%, respectively. MIC50 or MIC90 should only be used in an epidemiology context. For clarity, we recommend to always indicate the complete definition of an abbreviation in any report or article.
References
- B. Havlickova, V.A. Czaika, and M. Friedrich, "Epidemiological trends in skin mycoses worldwide", Mycoses, vol. 51, pp. 2-15, 2008. http://dx.doi.org/10.1111/j.1439-0507.2008.01606.x
- G.D. Brown, D.W. Denning, N.A.R. Gow, S.M. Levitz, M.G. Netea, and T.C. White, "Hidden Killers: Human Fungal Infections", Science Translational Medicine, vol. 4, 2012. http://dx.doi.org/10.1126/scitranslmed.3004404
- M. Dadar, R. Tiwari, K. Karthik, S. Chakraborty, Y. Shahali, and K. Dhama, "Candida albicans - Biology, molecular characterization, pathogenicity, and advances in diagnosis and control – An update", Microbial Pathogenesis, vol. 117, pp. 128-138, 2018. http://dx.doi.org/10.1016/j.micpath.2018.02.028
- A.K. Gupta, K.A. Foley, and S.G. Versteeg, "New Antifungal Agents and New Formulations Against Dermatophytes", Mycopathologia, vol. 182, pp. 127-141, 2016. http://dx.doi.org/10.1007/s11046-016-0045-0
- D. Farmakiotis, and D.P. Kontoyiannis, "Epidemiology of antifungal resistance in human pathogenic yeasts: current viewpoint and practical recommendations for management", International Journal of Antimicrobial Agents, vol. 50, pp. 318-324, 2017. http://dx.doi.org/10.1016/j.ijantimicag.2017.05.019
- S.R. Lockhart, E.L. Berkow, N. Chow, and R.M. Welsh, "Candida auris for the Clinical Microbiology Laboratory: Not Your Grandfather's Candida Species", Clinical Microbiology Newsletter, vol. 39, pp. 99-103, 2017. http://dx.doi.org/10.1016/j.clinmicnews.2017.06.003
- P.E. Verweij, A. Chowdhary, W.J.G. Melchers, and J.F. Meis, "Azole Resistance inAspergillus fumigatus: Can We Retain the Clinical Use of Mold-Active Antifungal Azoles?", Clinical Infectious Diseases, vol. 62, pp. 362-368, 2015. http://dx.doi.org/10.1093/cid/civ885
- A. Dongari-Bagtzoglou, "Pathogenesis of mucosal biofilm infections: challenges and progress", Expert Review of Anti-infective Therapy, vol. 6, pp. 201-208, 2008. http://dx.doi.org/10.1586/14787210.6.2.201
- C.A. Muzny, and J.R. Schwebke, "Biofilms: An Underappreciated Mechanism of Treatment Failure and Recurrence in Vaginal Infections: Table 1.", Clinical Infectious Diseases, vol. 61, pp. 601-606, 2015. http://dx.doi.org/10.1093/cid/civ353
- D. Davies, "Understanding biofilm resistance to antibacterial agents", Nature Reviews Drug Discovery, vol. 2, pp. 114-122, 2003. http://dx.doi.org/10.1038/nrd1008
- G. Morace, F. Perdoni, and E. Borghi, "Antifungal drug resistance in Candida species", Journal of Global Antimicrobial Resistance, vol. 2, pp. 254-259, 2014. http://dx.doi.org/10.1016/j.jgar.2014.09.002
- H. Van Acker, P. Van Dijck, and T. Coenye, "Molecular mechanisms of antimicrobial tolerance and resistance in bacterial and fungal biofilms", Trends in Microbiology, vol. 22, pp. 326-333, 2014. http://dx.doi.org/10.1016/j.tim.2014.02.001
- M. Sanguinetti, and B. Posteraro, "New approaches for antifungal susceptibility testing", Clinical Microbiology and Infection, vol. 23, pp. 931-934, 2017. http://dx.doi.org/10.1016/j.cmi.2017.03.025
- J. Sjollema, S.A. Zaat, V. Fontaine, M. Ramstedt, R. Luginbuehl, K. Thevissen, J. Li, H.C. van der Mei, and H.J. Busscher, "In vitro methods for the evaluation of antimicrobial surface designs", Acta Biomaterialia, vol. 70, pp. 12-24, 2018. http://dx.doi.org/10.1016/j.actbio.2018.02.001
- M. Arvanitis, J. Glavis-Bloom, and E. Mylonakis, "Invertebrate models of fungal infection", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1832, pp. 1378-1383, 2013. http://dx.doi.org/10.1016/j.bbadis.2013.03.008
- A. Espinel-Ingroff, and J. Turnidge, "The role of epidemiological cutoff values (ECVs/ECOFFs) in antifungal susceptibility testing and interpretation for uncommon yeasts and moulds", Revista Iberoamericana de Micología, vol. 33, pp. 63-75, 2016. http://dx.doi.org/10.1016/j.riam.2016.04.001
- . Clinical and Laboratory Standards Institute, "Reference method for broth dilution antifungal susceptibility testing of yeasts. 4th ed. CLSI standard M27.", Clinical and Laboratory Standards Institute, Wayne, PA., 2017.
- . Clinical and Laboratory Standards Institute, "Performance standards for antifungal susceptibility testing of yeasts. 1st ed. CLSI standard M60.", Clinical and Laboratory Standards Institute, Wayne, PA., 2017.
- . Clinical and Laboratory Standards Institute, "Reference method for broth dilution antifungal susceptibility testing of filamentous fungi. 3rd ed. CLSI standard M38.", Clinical and Laboratory Standards Institute, Wayne, PA., 2017.
- . Clinical and Laboratory Standards Institute, "Method for antifungal disk diffusion susceptibility testing of nondermatophyte filamentous fungi. 1st. ed. CLSI approved guideline M51.", Clinical and Laboratory Standards Institute, Wayne, PA., 2017.
- . Clinical and Laboratory Standards Institute, "Performance standards for antifungal susceptibility testing of filamentous fungi. 1st ed. CLSI standard M61.", Clinical and Laboratory Standards Institute, Wayne, PA., 2017.
- . Clinical and Laboratory Standards Institute, "Method for antifungal disk diffusionfor susceptibility testing of yeasts. 2nd ed. CLSI standard M 44.", Clinical and Laboratory Standards Institute, Wayne, PA., 2017.
- M. Arendrup, J. Meletiadis, J. Mouton, J. Guinea, M. Cuenca-Estrella, K. Lagrou, S. Howard, M. Arendrup, J. Meletiadis, S. Howard, J. Mouton, J. Guinea, K. Lagrou, S. Arikan-Akdagli, F. Barchiesi, P. Hamal, H. Järv, C. Lass-Flörl, M. Mares, T. Matos, K. Muehlethaler, T. Rogers, C. Torp Andersen, and P. Verweij, "EUCAST technical note on isavuconazole breakpoints for Aspergillus, itraconazole breakpoints for Candida and updates for the antifungal susceptibility testing method documents", Clinical Microbiology and Infection, vol. 22, pp. 571.e1-571.e4, 2016. http://dx.doi.org/10.1016/j.cmi.2016.01.017
- M.C. Arendrup, M. Cuenca-Estrella, C. Lass-Flörl, and W.W. Hope, "Breakpoints for antifungal agents: An update from EUCAST focussing on echinocandins against Candida spp. and triazoles against Aspergillus spp.", Drug Resistance Updates, vol. 16, pp. 81-95, 2013. http://dx.doi.org/10.1016/j.drup.2014.01.001
- M.C. Arendrup, A. Prakash, J. Meletiadis, C. Sharma, and A. Chowdhary, "Comparison of EUCAST and CLSI Reference Microdilution MICs of Eight Antifungal Compounds for Candida auris and Associated Tentative Epidemiological Cutoff Values", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.00485-17
- L.A. Walker, N.A. Gow, and C.A. Munro, "Fungal echinocandin resistance", Fungal Genetics and Biology, vol. 47, pp. 117-126, 2010. http://dx.doi.org/10.1016/j.fgb.2009.09.003
- N.B. Wertheimer, N. Stone, and J. Berman, "Ploidy dynamics and evolvability in fungi", Philosophical Transactions of the Royal Society B: Biological Sciences, vol. 371, pp. 20150461, 2016. http://dx.doi.org/10.1098/rstb.2015.0461
- E. Johnson, and M. Arendrup, " Susceptibility test methods: yeasts and filamentous fungi. ", In: Man. Clin. Microbiol. p 2255., 2015.
- . Clinical and Laboratory Standards Institute, "Principles and procedures for the development of epidemiological cutoff values for antifungal susceptibility testing. CLSI M57 document, 1st ed.", Clinical and Laboratory Standards Institute, Wayne, PA., 2016.
- . M59Ed2, "Epidemiological Cutoff Values for Antifungal Susceptibility Testing", Available: https://clsi.org/standards/products/microbiology/documents/m59/. Accessed 03.29.2018, 2018.
- A.C. Gerstein, A. Rosenberg, I. Hecht, and J. Berman, "diskImageR: quantification of resistance and tolerance to antimicrobial drugs using disk diffusion assays", Microbiology, vol. 162, pp. 1059-1068, 2016. http://dx.doi.org/10.1099/mic.0.000295
- J.R. Graybill, D.S. Burgess, and T.C. Hardin, "Key issues concerning fungistatic versus fungicidal drugs.", European journal of clinical microbiology & infectious diseases : official publication of the European Society of Clinical Microbiology, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9063673
- M.A. Pfaller, D.J. Sheehan, and J.H. Rex, "Determination of Fungicidal Activities against Yeasts and Molds: Lessons Learned from Bactericidal Testing and the Need for Standardization", Clinical Microbiology Reviews, vol. 17, pp. 268-280, 2004. http://dx.doi.org/10.1128/CMR.17.2.268-280.2004
- A.K. Gupta, B.E. Elewski, T. Rosen, B. Caldwell, D.M. Pariser, L.H. Kircik, N. Bhatia, and A. Tosti, "Onychomycosis: Strategies to Minimize Recurrence.", Journal of drugs in dermatology : JDD, 2016. http://www.ncbi.nlm.nih.gov/pubmed/26954312
- L.C. Whitney, and T. Bicanic, "Treatment Principles for Candida and Cryptococcus", Cold Spring Harbor Perspectives in Medicine, vol. 5, pp. a024158-a024158, 2014. http://dx.doi.org/10.1101/cshperspect.a024158
- J.D. Sobel, "Management of patients with recurrent vulvovaginal candidiasis.", Drugs, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12749733
- L.A. Walker, K.K. Lee, C.A. Munro, and N.A.R. Gow, "Caspofungin Treatment of Aspergillus fumigatus Results in ChsG-Dependent Upregulation of Chitin Synthesis and the Formation of Chitin-Rich Microcolonies", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 5932-5941, 2015. http://dx.doi.org/10.1128/AAC.00862-15
- E. Cantón, J. Pemán, A. Viudes, G. Quindós, M. Gobernado, and A. Espinel-Ingroff, "Minimum fungicidal concentrations of amphotericin B for bloodstream Candida species.", Diagnostic microbiology and infectious disease, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12663162
- G. Hazirolan, E. Canton, S. Sahin, and S. Arikan-Akdagli, "Head-to-Head Comparison of Inhibitory and Fungicidal Activities of Fluconazole, Itraconazole, Voriconazole, Posaconazole, and Isavuconazole against Clinical Isolates of Trichosporon asahii", Antimicrobial Agents and Chemotherapy, vol. 57, pp. 4841-4847, 2013. http://dx.doi.org/10.1128/AAC.00850-13
- M.E. Klepser, E.J. Wolfe, R.N. Jones, C.H. Nightingale, and M.A. Pfaller, "Antifungal pharmacodynamic characteristics of fluconazole and amphotericin B tested against Candida albicans.", Antimicrobial agents and chemotherapy, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9174207
- M.E. Klepser, E.J. Ernst, R.E. Lewis, M.E. Ernst, and M.A. Pfaller, "Influence of test conditions on antifungal time-kill curve results: proposal for standardized methods.", Antimicrobial agents and chemotherapy, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9593151
- B. Scorneaux, D. Angulo, K. Borroto-Esoda, M. Ghannoum, M. Peel, and S. Wring, "SCY-078 Is Fungicidal against Candida Species in Time-Kill Studies", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.01961-16
- E. Canton, J. Peman, D. Hervas, and A. Espinel-Ingroff, "Examination of the in vitro fungicidal activity of echinocandins against Candida lusitaniae by time-killing methods", Journal of Antimicrobial Chemotherapy, vol. 68, pp. 864-868, 2012. http://dx.doi.org/10.1093/jac/dks489
- S. Gil-Alonso, N. Jauregizar, E. Cantón, E. Eraso, and G. Quindós, "In Vitro Fungicidal Activities of Anidulafungin, Caspofungin, and Micafungin against Candida glabrata, Candida bracarensis, and Candida nivariensis Evaluated by Time-Kill Studies", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 3615-3618, 2015. http://dx.doi.org/10.1128/AAC.04474-14
- S. Tati, R. Li, S. Puri, R. Kumar, P. Davidow, and M. Edgerton, "Histatin 5-Spermidine Conjugates Have Enhanced Fungicidal Activity and Efficacy as a Topical Therapeutic for Oral Candidiasis", Antimicrobial Agents and Chemotherapy, vol. 58, pp. 756-766, 2014. http://dx.doi.org/10.1128/AAC.01851-13
- R. Kumar, S. Chadha, D. Saraswat, J.S. Bajwa, R.A. Li, H.R. Conti, and M. Edgerton, "Histatin 5 Uptake by Candida albicans Utilizes Polyamine Transporters Dur3 and Dur31 Proteins", Journal of Biological Chemistry, vol. 286, pp. 43748-43758, 2011. http://dx.doi.org/10.1074/jbc.M111.311175
- A. Espinel-Ingroff, A. Fothergill, J. Peter, M.G. Rinaldi, and T.J. Walsh, "Testing conditions for determination of minimum fungicidal concentrations of new and established antifungal agents for Aspergillus spp.: NCCLS collaborative study.", Journal of clinical microbiology, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12202554
- M.C.A. Leite, A.P.D.B. Bezerra, J.P.D. Sousa, F.Q.S. Guerra, and E.D.O. Lima, "Evaluation of Antifungal Activity and Mechanism of Action of Citral againstCandida albicans", Evidence-Based Complementary and Alternative Medicine, vol. 2014, pp. 1-9, 2014. http://dx.doi.org/10.1155/2014/378280
- S.P. Hawser, H. Norris, C.J. Jessup, and M.A. Ghannoum, "Comparison of a 2,3-bis(2-methoxy-4-nitro-5-sulfophenyl)-5-[(phenylamino)carbonyl]-2H-t etrazolium hydroxide (XTT) colorimetric method with the standardized National Committee for Clinical Laboratory Standards method of testing clinical yeast isolates for susceptibility to antifungal agents.", Journal of clinical microbiology, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9574729
- R. Tellier, M. Krajden, G.A. Grigoriew, and I. Campbell, "Innovative endpoint determination system for antifungal susceptibility testing of yeasts.", Antimicrobial agents and chemotherapy, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1416843
- D. Carmona-Gutierrez, M.A. Bauer, A. Zimmermann, A. Aguilera, N. Austriaco, K. Ayscough, R. Balzan, S. Bar-Nun, A. Barrientos, P. Belenky, M. Blondel, R.J. Braun, M. Breitenbach, W.C. Burhans, S. Buettner, D. Cavalieri, M. Chang, K.F. Cooper, M. Côrte-Real, V. Costa, C. Cullin, I. Dawes, J. Dengjel, M.B. Dickman, T. Eisenberg, B. Fahrenkrog, N. Fasel, K. Froehlich, A. Gargouri, S. Giannattasio, P. Goffrini, C.W. Gourlay, C.M. Grant, M.T. Greenwood, N. Guaragnella, T. Heger, J. Heinisch, E. Herker, J.M. Herrmann, S. Hofer, A. Jiménez-Ruiz, H. Jungwirth, K. Kainz, D.P. Kontoyiannis, P. Ludovico, S. Manon, E. Martegani, C. Mazzoni, L.A. Megeney, C. Meisinger, J. Nielsen, T. Nystroem, H.D. Osiewacz, T.F. Outeiro, H. Park, T. Pendl, D. Petranovic, S. Picot, P. Polčic, T. Powers, M. Ramsdale, M. Rinnerthaler, P. Rockenfeller, C. Ruckenstuhl, R. Schaffrath, M. Segovia, F.F. Severin, A. Sharon, S.J. Sigrist, C. Sommer-Ruck, M.J. Sousa, J.M. Thevelein, K. Thevissen, V. Titorenko, M.B. Toledano, M. Tuite, F. Voegtle, B. Westermann, J. Winderickx, S. Wissing, S. Woelfl, Z.J. Zhang, R.Y. Zhao, B. Zhou, L. Galluzzi, G. Kroemer, and F. Madeo, "Guidelines and recommendations on yeast cell death nomenclature", Microbial Cell, vol. 5, pp. 4-31, 2018. http://dx.doi.org/10.15698/mic2018.01.607
- M.C. Monteiro, M. de la Cruz, J. Cantizani, C. Moreno, J.R. Tormo, E. Mellado, J.R. De Lucas, F. Asensio, V. Valiante, A.A. Brakhage, J. Latgé, O. Genilloud, and F. Vicente, "A New Approach to Drug Discovery: High-Throughput Screening of Microbial Natural Extracts against Aspergillus fumigatus Using Resazurin", SLAS Discovery, vol. 17, pp. 542-549, 2012. http://dx.doi.org/10.1177/1087057111433459
- M.A. Pfaller, and A.L. Barry, "Evaluation of a novel colorimetric broth microdilution method for antifungal susceptibility testing of yeast isolates.", Journal of clinical microbiology, 1994. http://www.ncbi.nlm.nih.gov/pubmed/7989555
- B. Moriyama, S.A. Henning, S.R. Penzak, and T.J. Walsh, "The Postantifungal and Paradoxical Effects of Echinocandins Against Candida spp.", Future Microbiology, vol. 7, pp. 565-569, 2012. http://dx.doi.org/10.2217/fmb.12.31
- E. Cantón, A. Espinel-Ingroff, J. Pemán, and L. del Castillo, "In Vitro Fungicidal Activities of Echinocandins against Candida metapsilosis , C. orthopsilosis , and C. parapsilosis Evaluated by Time-Kill Studies", Antimicrobial Agents and Chemotherapy, vol. 54, pp. 2194-2197, 2010. http://dx.doi.org/10.1128/AAC.01538-09
- D.A. Stevens, T.C. White, D.S. Perlin, and C.P. Selitrennikoff, "Studies of the paradoxical effect of caspofungin at high drug concentrations", Diagnostic Microbiology and Infectious Disease, vol. 51, pp. 173-178, 2005. http://dx.doi.org/10.1016/j.diagmicrobio.2004.10.006
- C. Rueda, M. Cuenca-Estrella, and O. Zaragoza, "Paradoxical Growth of Candida albicans in the Presence of Caspofungin Is Associated with Multiple Cell Wall Rearrangements and Decreased Virulence", Antimicrobial Agents and Chemotherapy, vol. 58, pp. 1071-1083, 2014. http://dx.doi.org/10.1128/AAC.00946-13
- I. François, K. Thevissen, K. Pellens, E. Meert, J. Heeres, E. Freyne, E. Coesemans, M. Viellevoye, F. Deroose, S. Martinez Gonzalez, J. Pastor, D. Corens, L. Meerpoel, M. Borgers, J. Ausma, G. Dispersyn, and B. Cammue, "Design and Synthesis of a Series of Piperazine‐1‐carboxamidine Derivatives with Antifungal Activity Resulting from Accumulation of Endogenous Reactive Oxygen Species", ChemMedChem, vol. 4, pp. 1714-1721, 2009. http://dx.doi.org/10.1002/cmdc.200900249
- A.F. Feyaerts, L. Mathé, W. Luyten, S. De Graeve, K. Van Dyck, L. Broekx, and P. Van Dijck, "Essential oils and their components are a class of antifungals with potent vapour-phase-mediated anti-Candida activity", Scientific Reports, vol. 8, 2018. http://dx.doi.org/10.1038/s41598-018-22395-6
- L.J. Marcos-Zambrano, P. Escribano, C. Sánchez-Carrillo, E. Bouza, and J. Guinea, "Scope and frequency of fluconazole trailing assessed using EUCAST in invasiveCandidaspp. isolates", Medical Mycology, vol. 54, pp. 733-739, 2016. http://dx.doi.org/10.1093/mmy/myw033
- T. Chang, P. Lo, A. Wang, H. Lo, M. Ho, Y. Yang, and P. Lin, "Distribution and drug susceptibilities of Candida species causing candidemia from a medical center in central Taiwan", Journal of Infection and Chemotherapy, vol. 19, pp. 1065-1071, 2013. http://dx.doi.org/10.1007/s10156-013-0623-8
- E. Delarze, and D. Sanglard, "Defining the frontiers between antifungal resistance, tolerance and the concept of persistence", Drug Resistance Updates, vol. 23, pp. 12-19, 2015. http://dx.doi.org/10.1016/j.drup.2015.10.001
- T. Coenye, M. De Vos, D. Vandenbosch, and H. Nelis, "Factors influencing the trailing endpoint observed in Candida albicans susceptibility testing using the CLSI procedure", Clinical Microbiology and Infection, vol. 14, pp. 495-497, 2008. http://dx.doi.org/10.1111/j.1469-0691.2008.01956.x
- A. Rosenberg, I.V. Ene, M. Bibi, S. Zakin, E.S. Segal, N. Ziv, A.M. Dahan, A.L. Colombo, R.J. Bennett, and J. Berman, "Clearing the FoG: Antifungal tolerance is a subpopulation effect that is distinct from resistance and is associated with persistent candidemia", 2017. http://dx.doi.org/10.1101/206359
- M.D. LaFleur, Q. Qi, and K. Lewis, "Patients with Long-Term Oral Carriage Harbor High-Persister Mutants of Candida albicans", Antimicrobial Agents and Chemotherapy, vol. 54, pp. 39-44, 2010. http://dx.doi.org/10.1128/AAC.00860-09
- M.D. LaFleur, C.A. Kumamoto, and K. Lewis, "Candida albicans Biofilms Produce Antifungal-Tolerant Persister Cells", Antimicrobial Agents and Chemotherapy, vol. 50, pp. 3839-3846, 2006. http://dx.doi.org/10.1128/AAC.00684-06
- E. Sionov, H. Lee, Y.C. Chang, and K.J. Kwon-Chung, "Cryptococcus neoformans Overcomes Stress of Azole Drugs by Formation of Disomy in Specific Multiple Chromosomes", PLoS Pathogens, vol. 6, pp. e1000848, 2010. http://dx.doi.org/10.1371/journal.ppat.1000848
- E. Sionov, Y.C. Chang, and K.J. Kwon-Chung, "Azole Heteroresistance in Cryptococcus neoformans: Emergence of Resistant Clones with Chromosomal Disomy in the Mouse Brain during Fluconazole Treatment", Antimicrobial Agents and Chemotherapy, vol. 57, pp. 5127-5130, 2013. http://dx.doi.org/10.1128/AAC.00694-13
- E. Sionov, Y.C. Chang, H.M. Garraffo, and K.J. Kwon-Chung, "Heteroresistance to Fluconazole in Cryptococcus neoformans Is Intrinsic and Associated with Virulence", Antimicrobial Agents and Chemotherapy, vol. 53, pp. 2804-2815, 2009. http://dx.doi.org/10.1128/AAC.00295-09
- R. Ben-Ami, O. Zimmerman, T. Finn, S. Amit, A. Novikov, N. Wertheimer, M. Lurie-Weinberger, and J. Berman, "Heteroresistance to Fluconazole Is a Continuously Distributed Phenotype among Candida glabrata Clinical Strains Associated with In Vivo Persistence", mBio, vol. 7, 2016. http://dx.doi.org/10.1128/mBio.00655-16
- G. Ramage, and C. Williams, "The Clinical Importance of Fungal Biofilms", Advances in Applied Microbiology, pp. 27-83, 2013. http://dx.doi.org/10.1016/B978-0-12-407673-0.00002-3
- J. Nett, "The Host’s Reply to Candida Biofilm", Pathogens, vol. 5, pp. 33, 2016. http://dx.doi.org/10.3390/pathogens5010033
- M.A. Pfaller, and D.J. Diekema, "Epidemiology of Invasive Candidiasis: a Persistent Public Health Problem", Clinical Microbiology Reviews, vol. 20, pp. 133-163, 2007. http://dx.doi.org/10.1128/CMR.00029-06
- G. Ramage, J.P. MartÃnez, and J.L. López-Ribot, "Candidabiofilms on implanted biomaterials: a clinically significant problem", FEMS Yeast Research, vol. 6, pp. 979-986, 2006. http://dx.doi.org/10.1111/j.1567-1364.2006.00117.x
- C. Pierce, T. Vila, J. Romo, D. Montelongo-Jauregui, G. Wall, A. Ramasubramanian, and J. Lopez-Ribot, "The Candida albicans Biofilm Matrix: Composition, Structure and Function", Journal of Fungi, vol. 3, pp. 14, 2017. http://dx.doi.org/10.3390/jof3010014
- A. Beauvais, and J. Latgé, "Aspergillus Biofilm In Vitro and In Vivo", Microbiology Spectrum, vol. 3, 2015. http://dx.doi.org/10.1128/microbiolspec.MB-0017-2015
- E.G. Di Domenico, I. Cavallo, M. Guembe, G. Prignano, M.T. Gallo, V. Bordignon, G. D’Agosto, I. Sperduti, L. Toma, and F. Ensoli, "The clinical Biofilm Ring Test: a promising tool for the clinical assessment of biofilm-producing Candida species", FEMS Yeast Research, vol. 18, 2018. http://dx.doi.org/10.1093/femsyr/foy025
- G. Ramage, K. Vande Walle, B.L. Wickes, and J.L. López-Ribot, "Standardized method for in vitro antifungal susceptibility testing of Candida albicans biofilms.", Antimicrobial agents and chemotherapy, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11502517
- C.G. Pierce, P. Uppuluri, A.R. Tristan, F.L. Wormley, E. Mowat, G. Ramage, and J.L. Lopez-Ribot, "A simple and reproducible 96-well plate-based method for the formation of fungal biofilms and its application to antifungal susceptibility testing", Nature Protocols, vol. 3, pp. 1494-1500, 2008. http://dx.doi.org/10.1038/nprot.2008.141
- I. Vandecandelaere, H. Van Acker, and T. Coenye, "A Microplate-Based System as In Vitro Model of Biofilm Growth and Quantification", Methods in Molecular Biology, pp. 53-66, 2016. http://dx.doi.org/10.1007/978-1-4939-2854-5_5
- G. Ramage, "Comparing apples and oranges: considerations for quantifying candidal biofilms with XTT [2,3-bis(2-methoxy-4-nitro-5-sulfo-phenyl)-2H-tetrazolium-5-carboxanilide] and the need for standardized testing", Journal of Medical Microbiology, vol. 65, pp. 259-260, 2016. http://dx.doi.org/10.1099/jmm.0.000237
- G. Sass, H. Nazik, J. Penner, H. Shah, S.R. Ansari, K.V. Clemons, M. Groleau, A. Dietl, P. Visca, H. Haas, E. Déziel, and D.A. Stevens, "Studies of Pseudomonas aeruginosa Mutants Indicate Pyoverdine as the Central Factor in Inhibition of Aspergillus fumigatus Biofilm", Journal of Bacteriology, vol. 200, 2018. http://dx.doi.org/10.1128/JB.00345-17
- M.B. Lohse, M. Gulati, A. Valle Arevalo, A. Fishburn, A.D. Johnson, and C.J. Nobile, "Assessment and Optimizations of Candida albicans In Vitro Biofilm Assays", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.02749-16
- M. Feldman, J. Shenderovich, E. Lavy, M. Friedman, and D. Steinberg, "A Sustained-Release Membrane of Thiazolidinedione-8: Effect on Formation of a Candida/Bacteria Mixed Biofilm on Hydroxyapatite in a Continuous Flow Model", BioMed Research International, vol. 2017, pp. 1-9, 2017. http://dx.doi.org/10.1155/2017/3510124
- J. Shao, K. Lu, G. Tian, Y. Cui, Y. Yan, T. Wang, X. Zhang, and C. Wang, "Lab-scale preparations of Candida albicans and dual Candida albicans–Candida glabrata biofilms on the surface of medical-grade polyvinyl chloride (PVC) perfusion tube using a modified gravity-supported free-flow biofilm incubator (GS-FFBI)", Journal of Microbiological Methods, vol. 109, pp. 41-48, 2015. http://dx.doi.org/10.1016/j.mimet.2014.12.006
- A. McCall, and M. Edgerton, "Real-Time Approach to Flow Cell Imaging of Candida albicans Biofilm Development", Journal of Fungi, vol. 3, pp. 13, 2017. http://dx.doi.org/10.3390/jof3010013
- Y. Yawata, J. Nguyen, R. Stocker, and R. Rusconi, "Microfluidic Studies of Biofilm Formation in Dynamic Environments", Journal of Bacteriology, vol. 198, pp. 2589-2595, 2016. http://dx.doi.org/10.1128/JB.00118-16
- M. Gulati, C.L. Ennis, D.L. Rodriguez, and C.J. Nobile, "Visualization of Biofilm Formation in <em>Candida albicans</em> Using an Automated Microfluidic Device", Journal of Visualized Experiments, 2017. http://dx.doi.org/10.3791/56743
- A. Srinivasan, N.S. Torres, K.P. Leung, J.L. Lopez-Ribot, and A.K. Ramasubramanian, "nBio Chip, a Lab-on-a-Chip Platform of Mono- and Polymicrobial Biofilms for High-Throughput Downstream Applications", mSphere, vol. 2, 2017. http://dx.doi.org/10.1128/mSphere.00247-17
- A. Srinivasan, J.L. Lopez-Ribot, and A.K. Ramasubramanian, "<em>Candida albicans</em> Biofilm Chip (<em>Ca</em>BChip) for High-throughput Antifungal Drug Screening", Journal of Visualized Experiments, 2012. http://dx.doi.org/10.3791/3845
- A. Srinivasan, P. Uppuluri, J. Lopez-Ribot, and A.K. Ramasubramanian, "Development of a High-Throughput Candida albicans Biofilm Chip", PLoS ONE, vol. 6, pp. e19036, 2011. http://dx.doi.org/10.1371/journal.pone.0019036
- K. Vriens, T.L. Cools, P.J. Harvey, D.J. Craik, A. Braem, J. Vleugels, B.D. Coninck, B.P. Cammue, and K. Thevissen, "The radish defensins RsAFP1 and RsAFP2 act synergistically with caspofungin against Candida albicans biofilms", Peptides, vol. 75, pp. 71-79, 2016. http://dx.doi.org/10.1016/j.peptides.2015.11.001
- T.L. Cools, C. Struyfs, J.W. Drijfhout, S. Kucharíková, C. Lobo Romero, P. Van Dijck, M.H.S. Ramada, C. Bloch, B.P.A. Cammue, and K. Thevissen, "A Linear 19-Mer Plant Defensin-Derived Peptide Acts Synergistically with Caspofungin against Candida albicans Biofilms", Frontiers in Microbiology, vol. 8, 2017. http://dx.doi.org/10.3389/fmicb.2017.02051
- N. Delattin, K. De Brucker, K. De Cremer, B. P.A. Cammue, and K. Thevissen, "Antimicrobial Peptides as a Strategy to Combat Fungal Biofilms", Current Topics in Medicinal Chemistry, vol. 17, pp. 604-612, 2016. http://dx.doi.org/10.2174/1568026616666160713142228
- S. Silva, C. Rodrigues, D. Araújo, M. Rodrigues, and M. Henriques, "Candida Species Biofilms’ Antifungal Resistance", Journal of Fungi, vol. 3, pp. 8, 2017. http://dx.doi.org/10.3390/jof3010008
- L. Mathé, and P. Van Dijck, "Recent insights into Candida albicans biofilm resistance mechanisms", Current Genetics, vol. 59, pp. 251-264, 2013. http://dx.doi.org/10.1007/s00294-013-0400-3
- T. Coenye, D. Goeres, F. Van Bambeke, and T. Bjarnsholt, "Should standardized susceptibility testing for microbial biofilms be introduced in clinical practice?", Clinical Microbiology and Infection, vol. 24, pp. 570-572, 2018. http://dx.doi.org/10.1016/j.cmi.2018.01.003
- K. Thevissen, "How Promising are Combinatorial Drug Strategies in Combating Candida Albicans Biofilms?", Future Medicinal Chemistry, vol. 8, pp. 1383-1385, 2016. http://dx.doi.org/10.4155/fmc-2016-0127
- J.A. Hill, and L.E. Cowen, "Using Combination Therapy to Thwart Drug Resistance", Future Microbiology, vol. 10, pp. 1719-1726, 2015. http://dx.doi.org/10.2217/fmb.15.68
- N. Delattin, K. De Brucker, K. Vandamme, E. Meert, A. Marchand, P. Chaltin, B.P.A. Cammue, and K. Thevissen, "Repurposing as a means to increase the activity of amphotericin B and caspofungin against Candida albicans biofilms", Journal of Antimicrobial Chemotherapy, vol. 69, pp. 1035-1044, 2013. http://dx.doi.org/10.1093/jac/dkt449
- K. De Cremer, N. Delattin, K. De Brucker, A. Peeters, S. Kucharíková, E. Gerits, N. Verstraeten, J. Michiels, P. Van Dijck, B.P.A. Cammue, and K. Thevissen, "Oral Administration of the Broad-Spectrum Antibiofilm Compound Toremifene Inhibits Candida albicans and Staphylococcus aureus Biofilm Formation In Vivo", Antimicrobial Agents and Chemotherapy, vol. 58, pp. 7606-7610, 2014. http://dx.doi.org/10.1128/AAC.03869-14
- A.M. Troskie, M. Rautenbach, N. Delattin, J.A. Vosloo, M. Dathe, B.P.A. Cammue, and K. Thevissen, "Synergistic Activity of the Tyrocidines, Antimicrobial Cyclodecapeptides from Bacillus aneurinolyticus, with Amphotericin B and Caspofungin against Candida albicans Biofilms", Antimicrobial Agents and Chemotherapy, vol. 58, pp. 3697-3707, 2014. http://dx.doi.org/10.1128/AAC.02381-14
- K. Vriens, T.L. Cools, P.J. Harvey, D.J. Craik, P. Spincemaille, D. Cassiman, A. Braem, J. Vleugels, P.H. Nibbering, J.W. Drijfhout, B. De Coninck, B.P.A. Cammue, and K. Thevissen, "Synergistic Activity of the Plant Defensin HsAFP1 and Caspofungin against Candida albicans Biofilms and Planktonic Cultures", PLOS ONE, vol. 10, pp. e0132701, 2015. http://dx.doi.org/10.1371/journal.pone.0132701
- K. De Cremer, E. Lanckacker, T.L. Cools, M. Bax, K. De Brucker, P. Cos, B.P.A. Cammue, and K. Thevissen, "Artemisinins, New Miconazole Potentiators Resulting in Increased Activity against Candida albicans Biofilms", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 421-426, 2015. http://dx.doi.org/10.1128/AAC.04229-14
- L.V. Silva, M. Sanguinetti, P. Vandeputte, R. Torelli, B. Rochat, and D. Sanglard, "Milbemycins: More than Efflux Inhibitors for Fungal Pathogens", Antimicrobial Agents and Chemotherapy, vol. 57, pp. 873-886, 2013. http://dx.doi.org/10.1128/AAC.02040-12
- V. Valiante, M.C. Monteiro, J. Martín, R. Altwasser, N. El Aouad, I. González, O. Kniemeyer, E. Mellado, S. Palomo, N. de Pedro, I. Pérez-Victoria, J.R. Tormo, F. Vicente, F. Reyes, O. Genilloud, and A.A. Brakhage, "Hitting the Caspofungin Salvage Pathway of Human-Pathogenic Fungi with the Novel Lasso Peptide Humidimycin (MDN-0010)", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 5145-5153, 2015. http://dx.doi.org/10.1128/AAC.00683-15
- F.C. Odds, "Synergy, antagonism, and what the chequerboard puts between them", Journal of Antimicrobial Chemotherapy, vol. 52, pp. 1-1, 2003. http://dx.doi.org/10.1093/jac/dkg301
- D.T.A. Te Dorsthorst, P.E. Verweij, J.F.G.M. Meis, N.C. Punt, and J.W. Mouton, "Comparison of fractional inhibitory concentration index with response surface modeling for characterization of in vitro interaction of antifungals against itraconazole-susceptible and -resistant Aspergillus fumigatus isolates.", Antimicrobial agents and chemotherapy, 2002. http://www.ncbi.nlm.nih.gov/pubmed/11850251
- P. Wu, and D.W. Grainger, "Drug/device combinations for local drug therapies and infection prophylaxis", Biomaterials, vol. 27, pp. 2450-2467, 2006. http://dx.doi.org/10.1016/j.biomaterials.2005.11.031
- C.D. Klink, M. Binnebösel, A. Lambertz, H.P. Alizai, A. Roeth, J. Otto, U. Klinge, U.P. Neumann, and K. Junge, "In vitro and in vivo characteristics of gentamicin‐supplemented polyvinylidenfluoride mesh materials", Journal of Biomedical Materials Research Part A, vol. 100A, pp. 1195-1202, 2012. http://dx.doi.org/10.1002/jbm.a.34066
- S.Y. Wong, J.S. Moskowitz, J. Veselinovic, R.A. Rosario, K. Timachova, M.R. Blaisse, R.C. Fuller, A.M. Klibanov, and P.T. Hammond, "Dual Functional Polyelectrolyte Multilayer Coatings for Implants: Permanent Microbicidal Base with Controlled Release of Therapeutic Agents", Journal of the American Chemical Society, vol. 132, pp. 17840-17848, 2010. http://dx.doi.org/10.1021/ja106288c
- T.F. Moriarty, D.W. Grainger, and R.G. Richards, "Challenges in linking preclinical anti-microbial research strategies with clinical outcomes for device-associated infections.", European cells & materials, 2014. http://www.ncbi.nlm.nih.gov/pubmed/25214018
- E. Verraedt, A. Braem, A. Chaudhari, K. Thevissen, E. Adams, L. Van Mellaert, B.P. Cammue, J. Duyck, J. Anné, J. Vleugels, and J.A. Martens, "Controlled release of chlorhexidine antiseptic from microporous amorphous silica applied in open porosity of an implant surface", International Journal of Pharmaceutics, vol. 419, pp. 28-32, 2011. http://dx.doi.org/10.1016/j.ijpharm.2011.06.053
- M. Klevay, A. Ebinger, D. Diekema, S. Messer, R. Hollis, and M. Pfaller, "DiskDiffusion Testing Using Candida sp. Colonies Taken Directlyfrom CHROMagar Candida Medium May Decrease Time Required To ObtainResults", Journal of Clinical Microbiology, vol. 43, pp. 3497-3499, 2005. http://dx.doi.org/10.1128/jcm.43.7.3497-3499.2005
- B.S. Necula, L.E. Fratila-Apachitei, S.A. Zaat, I. Apachitei, and J. Duszczyk, "In vitro antibacterial activity of porous TiO2–Ag composite layers against methicillin-resistant Staphylococcus aureus", Acta Biomaterialia, vol. 5, pp. 3573-3580, 2009. http://dx.doi.org/10.1016/j.actbio.2009.05.010
- M. van de Lagemaat, A. Grotenhuis, B. van de Belt-Gritter, S. Roest, T.J. Loontjens, H.J. Busscher, H.C. van der Mei, and Y. Ren, "Comparison of methods to evaluate bacterial contact-killing materials", Acta Biomaterialia, vol. 59, pp. 139-147, 2017. http://dx.doi.org/10.1016/j.actbio.2017.06.042
- J. Sjollema, R.J. Dijkstra, C. Abeln, H.C. van der Mei, D. van Asseldonk, and H.J. Busscher, "On-demand antimicrobial release from a temperature-sensitive polymer — Comparison with ad libitum release from central venous catheters", Journal of Controlled Release, vol. 188, pp. 61-66, 2014. http://dx.doi.org/10.1016/j.jconrel.2014.06.013
- J. Sjollema, H. Keul, H. van der Mei, R. Dijkstra, M. Rustema‐Abbing, J. de Vries, T. Loontjens, T. Dirks, and H. Busscher, "A Trifunctional, Modular Biomaterial Coating: Nonadhesive to Bacteria, Chlorhexidine‐Releasing and Tissue‐Integrating", Macromolecular Bioscience, vol. 17, 2016. http://dx.doi.org/10.1002/mabi.201600336
- R.T.C. Cleophas, M. Riool, H.(.C. Quarles van Ufford, S.A.J. Zaat, J.A.W. Kruijtzer, and R.M.J. Liskamp, "Convenient Preparation of Bactericidal Hydrogels by Covalent Attachment of Stabilized Antimicrobial Peptides Using Thiol–ene Click Chemistry", ACS Macro Letters, vol. 3, pp. 477-480, 2014. http://dx.doi.org/10.1021/mz5001465
- A. Héquet, V. Humblot, J. Berjeaud, and C. Pradier, "Optimized grafting of antimicrobial peptides on stainless steel surface and biofilm resistance tests", Colloids and Surfaces B: Biointerfaces, vol. 84, pp. 301-309, 2011. http://dx.doi.org/10.1016/j.colsurfb.2011.01.012
- P. Uppuluri, A.K. Chaturvedi, and J.L. Lopez-Ribot, "Design of a Simple Model of Candida albicans Biofilms Formed under Conditions of Flow: Development, Architecture, and Drug Resistance", Mycopathologia, vol. 168, pp. 101-109, 2009. http://dx.doi.org/10.1007/s11046-009-9205-9
- T.E. Andersen, P. Kingshott, Y. Palarasah, M. Benter, M. Alei, and H.J. Kolmos, "A flow chamber assay for quantitative evaluation of bacterial surface colonization used to investigate the influence of temperature and surface hydrophilicity on the biofilm forming capacity of uropathogenic Escherichia coli", Journal of Microbiological Methods, vol. 81, pp. 135-140, 2010. http://dx.doi.org/10.1016/j.mimet.2010.02.009
- I.B. Gomes, A. Meireles, A.L. Gonçalves, D.M. Goeres, J. Sjollema, L.C. Simões, and M. Simões, "Standardized reactors for the study of medical biofilms: a review of the principles and latest modifications", Critical Reviews in Biotechnology, vol. 38, pp. 657-670, 2017. http://dx.doi.org/10.1080/07388551.2017.1380601
- T. Coenye, K. De Prijck, B. De Wever, and H. Nelis, "Use of the modified Robbins device to study thein vitrobiofilm removal efficacy of NitrAdine™, a novel disinfecting formula for the maintenance of oral medical devices", Journal of Applied Microbiology, vol. 105, pp. 733-740, 2008. http://dx.doi.org/10.1111/j.1365-2672.2008.03784.x
- H.J. Busscher, H.C. van der Mei, G. Subbiahdoss, P.C. Jutte, J.J.A.M. van den Dungen, S.A.J. Zaat, M.J. Schultz, and D.W. Grainger, "Biomaterial-Associated Infection: Locating the Finish Line in the Race for the Surface", Science Translational Medicine, vol. 4, 2012. http://dx.doi.org/10.1126/scitranslmed.3004528
- K.H. Bleicher, H. Böhm, K. Müller, and A.I. Alanine, "Hit and lead generation: beyond high-throughput screening", Nature Reviews Drug Discovery, vol. 2, pp. 369-378, 2003. http://dx.doi.org/10.1038/nrd1086
- W. Gu, Q. Yu, C. Yu, and S. Sun, "In vivo activity of fluconazole/tetracycline combinations in Galleria mellonella with resistant Candida albicans infection", Journal of Global Antimicrobial Resistance, vol. 13, pp. 74-80, 2018. http://dx.doi.org/10.1016/j.jgar.2017.11.011
- L. Ames, S. Duxbury, B. Pawlowska, H. Ho, K. Haynes, and S. Bates, "Galleria mellonella as a host model to study Candida glabrata virulence and antifungal efficacy", Virulence, vol. 8, pp. 1909-1917, 2017. http://dx.doi.org/10.1080/21505594.2017.1347744
- B. Aneja, M. Irfan, C. Kapil, M.A. Jairajpuri, R. Maguire, K. Kavanagh, M.M.A. Rizvi, N. Manzoor, A. Azam, and M. Abid, "Effect of novel triazole–amino acid hybrids on growth and virulence of Candida species: in vitro and in vivo studies", Organic & Biomolecular Chemistry, vol. 14, pp. 10599-10619, 2016. http://dx.doi.org/10.1039/C6OB01718E
- M. Lu, C. Yu, X. Cui, J. Shi, L. Yuan, and S. Sun, "Gentamicin synergises with azoles against drug-resistant Candida albicans", International Journal of Antimicrobial Agents, vol. 51, pp. 107-114, 2018. http://dx.doi.org/10.1016/j.ijantimicag.2017.09.012
- A.C.R. Souza, B.B. Fuchs, H.M.S. Pinhati, R.A. Siqueira, F. Hagen, J.F. Meis, E. Mylonakis, and A.L. Colombo, "Candida parapsilosis Resistance to Fluconazole: Molecular Mechanisms and In Vivo Impact in Infected Galleria mellonella Larvae", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 6581-6587, 2015. http://dx.doi.org/10.1128/AAC.01177-15
- F. Lopez-Moya, M.F. Colom-Valiente, P. Martinez-Peinado, J.E. Martinez-Lopez, E. Puelles, J.M. Sempere-Ortells, and L.V. Lopez-Llorca, "Carbon and nitrogen limitation increase chitosan antifungal activity in Neurospora crassa and fungal human pathogens", Fungal Biology, vol. 119, pp. 154-169, 2015. http://dx.doi.org/10.1016/j.funbio.2014.12.003
- E. Maurer, N. Browne, C. Surlis, E. Jukic, P. Moser, K. Kavanagh, C. Lass-Flörl, and U. Binder, "Galleria mellonellaas a host model to studyAspergillus terreusvirulence and amphotericin B resistance", Virulence, vol. 6, pp. 591-598, 2015. http://dx.doi.org/10.1080/21505594.2015.1045183
- Q. Favre-Godal, S. Dorsaz, E.F. Queiroz, C. Conan, L. Marcourt, B.P.E. Wardojo, F. Voinesco, A. Buchwalder, K. Gindro, D. Sanglard, and J. Wolfender, "Comprehensive approach for the detection of antifungal compounds using a susceptible strain of Candida albicans and confirmation of in vivo activity with the Galleria mellonella model", Phytochemistry, vol. 105, pp. 68-78, 2014. http://dx.doi.org/10.1016/j.phytochem.2014.06.004
- D. Li, L. Deng, G. Hu, L. Zhao, D. Hu, Y. Jiang, and Y. Wang, "Using Galleria mellonella-Candida albicans infection model to evaluate antifungal agents.", Biological & pharmaceutical bulletin, 2013. http://www.ncbi.nlm.nih.gov/pubmed/23995660
- L. Scorzoni, M.P. de Lucas, A.C. Mesa-Arango, A.M. Fusco-Almeida, E. Lozano, M. Cuenca-Estrella, M.J. Mendes-Giannini, and O. Zaragoza, "Antifungal Efficacy during Candida krusei Infection in Non-Conventional Models Correlates with the Yeast In Vitro Susceptibility Profile", PLoS ONE, vol. 8, pp. e60047, 2013. http://dx.doi.org/10.1371/journal.pone.0060047
- A.C. Mesa-Arango, A. Forastiero, L. Bernal-Martínez, M. Cuenca-Estrella, E. Mellado, and O. Zaragoza, "The non-mammalian hostGalleria mellonellacan be used to study the virulence of the fungal pathogenCandida tropicalisand the efficacy of antifungal drugs during infection by this pathogenic yeast", Medical Mycology, vol. 51, pp. 461-472, 2013. http://dx.doi.org/10.3109/13693786.2012.737031
- R. Rowan, C. Moran, M. McCann, and K. Kavanagh, "Use of Galleria mellonella larvae to evaluate the in vivo anti-fungal activity of [Ag2(mal)(phen)3]", BioMetals, vol. 22, pp. 461-467, 2008. http://dx.doi.org/10.1007/s10534-008-9182-3
- E. Delarze, F. Ischer, D. Sanglard, and A.T. Coste, "Adaptation of aGaussia princepsLuciferase reporter system inCandida albicansforin vivodetection in theGalleria mellonellainfection model", Virulence, vol. 6, pp. 684-693, 2015. http://dx.doi.org/10.1080/21505594.2015.1081330
- A. Forastiero, L. Bernal-Martínez, E. Mellado, E. Cendejas, and A. Gomez-Lopez, "In vivo efficacy of voriconazole and posaconazole therapy in a novel invertebrate model of Aspergillus fumigatus infection", International Journal of Antimicrobial Agents, vol. 46, pp. 511-517, 2015. http://dx.doi.org/10.1016/j.ijantimicag.2015.07.007
- D. Ben Yaakov, A. Rivkin, G. Mircus, N. Albert, A. Dietl, D. Kovalerchick, S. Carmeli, H. Haas, D.P. Kontoyiannis, and N. Osherov, "Identification and characterization of haemofungin, a novel antifungal compound that inhibits the final step of haem biosynthesis", Journal of Antimicrobial Chemotherapy, vol. 71, pp. 946-952, 2016. http://dx.doi.org/10.1093/jac/dkv446
- D. Ben Yaakov, Y. Shadkchan, N. Albert, D.P. Kontoyiannis, and N. Osherov, "The quinoline bromoquinol exhibits broad-spectrum antifungal activity and induces oxidative stress and apoptosis in Aspergillus fumigatus", Journal of Antimicrobial Chemotherapy, vol. 72, pp. 2263-2272, 2017. http://dx.doi.org/10.1093/jac/dkx117
- L. Alcazar-Fuoli, M. Buitrago, A. Gomez-Lopez, and E. Mellado, "An alternative host model of a mixed fungal infection by azole susceptible and resistantAspergillusspp strains", Virulence, vol. 6, pp. 376-384, 2015. http://dx.doi.org/10.1080/21505594.2015.1025192
- A. Gomez-Lopez, A. Forastiero, E. Cendejas-Bueno, L. Gregson, E. Mellado, S. Howard, J. Livermore, W. Hope, and M. Cuenca-Estrella, "An invertebrate model to evaluate virulence in Aspergillus fumigatus: The role of azole resistance", Medical Mycology, vol. 52, pp. 311-319, 2014. http://dx.doi.org/10.1093/mmy/myt022
- A.C. Palanco, J.D. Lacorte Singulani, C.B. Costa-Orlandi, F.P. Gullo, N.M. Strohmayer Lourencetti, P.C. Gomes, G.M. Ayusso, L.A. Dutra, V.D. Silva Bolzani, L.O. Regasini, M.J. Soares Mendes-Giannini, and A.M. Fusco-Almeida, "Activity of 3′-Hydroxychalcone Against Cryptococcus Gattii and Toxicity, and Efficacy in Alternative Animal Models", Future Microbiology, vol. 12, pp. 1123-1134, 2017. http://dx.doi.org/10.2217/fmb-2017-0062
- N.P. de Sá, P.P. de Barros, J.C. Junqueira, J.A. Vaz, R.B. de Oliveira, C.A. Rosa, D.A. Santos, and S. Johann, "Thiazole derivatives act on virulence factors ofCryptococcusspp.", Medical Mycology, vol. 57, pp. 84-91, 2018. http://dx.doi.org/10.1093/mmy/myx158
- F. Sangalli-Leite, L. Scorzoni, A.C. Alves de Paula e Silva, J.D.F. da Silva, H.C. de Oliveira, J. de Lacorte Singulani, F.P. Gullo, R. Moraes da Silva, L.O. Regasini, D.H. Siqueira da Silva, V. da Silva Bolzani, A.M. Fusco-Almeida, and M.J. Soares Mendes-Giannini, "Synergistic effect of pedalitin and amphotericin B against Cryptococcus neoformans by in vitro and in vivo evaluation", International Journal of Antimicrobial Agents, vol. 48, pp. 504-511, 2016. http://dx.doi.org/10.1016/j.ijantimicag.2016.07.025
- K. Vu, and A. Gelli, "Astemizole and an analogue promote fungicidal activity of fluconazole against Cryptococcus neoformans var. grubii and Cryptococcus gattii", Medical Mycology, pp. 1-8, 2009. http://dx.doi.org/10.1080/13693780903081968
- E. Mylonakis, R. Moreno, J.B. El Khoury, A. Idnurm, J. Heitman, S.B. Calderwood, F.M. Ausubel, and A. Diener, "Galleria mellonella as a Model System To Study Cryptococcus neoformans Pathogenesis", Infection and Immunity, vol. 73, pp. 3842-3850, 2005. http://dx.doi.org/10.1128/IAI.73.7.3842-3850.2005
- W. Kloezen, F. Parel, R. Brüggemann, K. Asouit, M. Helvert-van Poppel, A. Fahal, J. Mouton, and W. van de Sande, "Amphotericin B and terbinafine but not the azoles prolong survival in Galleria mellonella larvae infected with Madurella mycetomatis", Medical Mycology, vol. 56, pp. 469-478, 2017. http://dx.doi.org/10.1093/mmy/myx064
- J. de Lacorte Singulani, L. Scorzoni, A.C.A. de Paula e Silva, A.M. Fusco-Almeida, and M.J.S. Mendes-Giannini, "Evaluation of the efficacy of antifungal drugs against Paracoccidioides brasiliensis and Paracoccidioides lutzii in a Galleria mellonella model", International Journal of Antimicrobial Agents, vol. 48, pp. 292-297, 2016. http://dx.doi.org/10.1016/j.ijantimicag.2016.05.012
- W. Kloezen, M. van Helvert-van Poppel, A.H. Fahal, and W.W.J. van de Sande, "A Madurella mycetomatis Grain Model in Galleria mellonella Larvae", PLOS Neglected Tropical Diseases, vol. 9, pp. e0003926, 2015. http://dx.doi.org/10.1371/journal.pntd.0003926
- M. Mariné, V.L.P. Bom, P.A. de Castro, L.K. Winkelstroter, L.N. Ramalho, N.A. Brown, and G.H. Goldman, "The development of animal infection models and antifungal efficacy assays against clinical isolates ofTrichosporon asahii,T. asteroidesandT. inkin", Virulence, vol. 6, pp. 476-486, 2015. http://dx.doi.org/10.1080/21505594.2015.1020273
- R.J. Bastidas, C.A. Shertz, S.C. Lee, J. Heitman, and M.E. Cardenas, "Rapamycin Exerts Antifungal Activity In Vitro and In Vivo against Mucor circinelloides via FKBP12-Dependent Inhibition of Tor", Eukaryotic Cell, vol. 11, pp. 270-281, 2012. http://dx.doi.org/10.1128/EC.05284-11
- J.J. Coleman, M. Muhammed, P.V. Kasperkovitz, J.M. Vyas, and E. Mylonakis, "Fusarium pathogenesis investigated using Galleria mellonella as a heterologous host", Fungal Biology, vol. 115, pp. 1279-1289, 2011. http://dx.doi.org/10.1016/j.funbio.2011.09.005
- R. Uchida, S. Namiguchi, H. Ishijima, and H. Tomoda, "Therapeutic effects of three trichothecenes in the silkworm infection assay with Candida albicans.", Drug discoveries & therapeutics, 2016. http://www.ncbi.nlm.nih.gov/pubmed/26971555
- I. Nakamura, R. Kanasaki, K. Yoshikawa, S. Furukawa, A. Fujie, H. Hamamoto, and K. Sekimizu, "Discovery of a new antifungal agent ASP2397 using a silkworm model of Aspergillus fumigatus infection", The Journal of Antibiotics, vol. 70, pp. 41-44, 2016. http://dx.doi.org/10.1038/ja.2016.106
- Y. Matsumoto, M. Ishii, K. Shimizu, S. Kawamoto, and K. Sekimizu, "A Silkworm Infection Model to Evaluate Antifungal Drugs for Cryptococcosis", Medical Mycology Journal, vol. 58, pp. E131-E137, 2017. http://dx.doi.org/10.3314/mmj.17.016
- M. Ishii, Y. Matsumoto, and K. Sekimizu, "Usefulness of silkworm as a host animal for understanding pathogenicity of Cryptococcus neoformans.", Drug discoveries & therapeutics, 2016. http://www.ncbi.nlm.nih.gov/pubmed/26902902
- Y. Matsumoto, S. Miyazaki, D. Fukunaga, K. Shimizu, S. Kawamoto, and K. Sekimizu, "Quantitative evaluation of cryptococcal pathogenesis and antifungal drugs using a silkworm infection model with Cryptococcus neoformans", Journal of Applied Microbiology, vol. 112, pp. 138-146, 2011. http://dx.doi.org/10.1111/j.1365-2672.2011.05186.x
- M. Muhammed, M. Arvanitis, and E. Mylonakis, "Whole animal HTS of small molecules for antifungal compounds", Expert Opinion on Drug Discovery, vol. 11, pp. 177-184, 2015. http://dx.doi.org/10.1517/17460441.2016.1122591
- L. Sun, K. Liao, and C. Hang, "Caffeic acid phenethyl ester synergistically enhances the antifungal activity of fluconazole against resistant Candida albicans", Phytomedicine, vol. 40, pp. 55-58, 2018. http://dx.doi.org/10.1016/j.phymed.2017.12.033
- G.A. Subramenium, T.K. Swetha, P.M. Iyer, K. Balamurugan, and S.K. Pandian, "5-hydroxymethyl-2-furaldehyde from marine bacterium Bacillus subtilis inhibits biofilm and virulence of Candida albicans", Microbiological Research, vol. 207, pp. 19-32, 2018. http://dx.doi.org/10.1016/j.micres.2017.11.002
- H. Mohammad, N.H. Elghazawy, H.E. Eldesouky, Y.A. Hegazy, W. Younis, L. Avrimova, T. Hazbun, R.K. Arafa, and M.N. Seleem, "Discovery of a Novel Dibromoquinoline Compound Exhibiting Potent Antifungal and Antivirulence Activity That Targets Metal Ion Homeostasis", ACS Infectious Diseases, vol. 4, pp. 403-414, 2018. http://dx.doi.org/10.1021/acsinfecdis.7b00215
- J.D.L. Singulani, L. Scorzoni, P.C. Gomes, A.C. Nazaré, C.R. Polaquini, L.O. Regasini, A.M. Fusco-Almeida, and M.J.S. Mendes-Giannini, "Activity of Gallic Acid and Its Ester Derivatives in Caenorhabditis Elegans and Zebrafish ( Danio Rerio ) Models", Future Medicinal Chemistry, vol. 9, pp. 1863-1872, 2017. http://dx.doi.org/10.4155/fmc-2017-0096
- S. Thangamani, M. Maland, H. Mohammad, P.E. Pascuzzi, L. Avramova, C.M. Koehler, T.R. Hazbun, and M.N. Seleem, "Repurposing Approach Identifies Auranofin with Broad Spectrum Antifungal Activity That Targets Mia40-Erv1 Pathway", Frontiers in Cellular and Infection Microbiology, vol. 7, 2017. http://dx.doi.org/10.3389/fcimb.2017.00004
- C. Shu, L. Sun, and W. Zhang, "Thymol has antifungal activity against Candida albicans during infection and maintains the innate immune response required for function of the p38 MAPK signaling pathway in Caenorhabditis elegans", Immunologic Research, vol. 64, pp. 1013-1024, 2016. http://dx.doi.org/10.1007/s12026-016-8785-y
- Y. Li, W. Chang, M. Zhang, Z. Ying, and H. Lou, "Natural product solasodine-3-O-β-D-glucopyranoside inhibits the virulence factors ofCandida albicans", FEMS Yeast Research, vol. 15, pp. fov060, 2015. http://dx.doi.org/10.1093/femsyr/fov060
- L. Sun, K. Liao, and D. Wang, "Effects of Magnolol and Honokiol on Adhesion, Yeast-Hyphal Transition, and Formation of Biofilm by Candida albicans", PLOS ONE, vol. 10, pp. e0117695, 2015. http://dx.doi.org/10.1371/journal.pone.0117695
- G. Vediyappan, V. Dumontet, F. Pelissier, and C. d’Enfert, "Gymnemic Acids Inhibit Hyphal Growth and Virulence in Candida albicans", PLoS ONE, vol. 8, pp. e74189, 2013. http://dx.doi.org/10.1371/journal.pone.0074189
- I. Okoli, J.J. Coleman, E. Tempakakis, W.F. An, E. Holson, F. Wagner, A.L. Conery, J. Larkins-Ford, G. Wu, A. Stern, F.M. Ausubel, and E. Mylonakis, "Identification of Antifungal Compounds Active against Candida albicans Using an Improved High-Throughput Caenorhabditis elegans Assay", PLoS ONE, vol. 4, pp. e7025, 2009. http://dx.doi.org/10.1371/journal.pone.0007025
- E. Tampakakis, I. Okoli, and E. Mylonakis, "A C. elegans-based, whole animal, in vivo screen for the identification of antifungal compounds", Nature Protocols, vol. 3, pp. 1925-1931, 2008. http://dx.doi.org/10.1038/nprot.2008.193
- J. Breger, B.B. Fuchs, G. Aperis, T.I. Moy, F.M. Ausubel, and E. Mylonakis, "Antifungal Chemical Compounds Identified Using a C. elegans Pathogenicity Assay", PLoS Pathogens, vol. 3, pp. e18, 2007. http://dx.doi.org/10.1371/journal.ppat.0030018
- S. Thangamani, H.E. Eldesouky, H. Mohammad, P.E. Pascuzzi, L. Avramova, T.R. Hazbun, and M.N. Seleem, "Ebselen exerts antifungal activity by regulating glutathione (GSH) and reactive oxygen species (ROS) production in fungal cells", Biochimica et Biophysica Acta (BBA) - General Subjects, vol. 1861, pp. 3002-3010, 2017. http://dx.doi.org/10.1016/j.bbagen.2016.09.029
- R.D.A. Cordeiro, A.J.D.J. Evangelista, R. Serpa, F.J.D.F. Marques, C.V.S.D. Melo, J.S.D. Oliveira, J.D.S. Franco, L.P.D. Alencar, T.D.J.P.G. Bandeira, R.S.N. Brilhante, J.J.C. Sidrim, and M.F.G. Rocha, "Inhibition of heat-shock protein 90 enhances the susceptibility to antifungals and reduces the virulence of Cryptococcus neoformans/Cryptococcus gattii species complex", Microbiology, vol. 162, pp. 309-317, 2016. http://dx.doi.org/10.1099/mic.0.000222
- X. Huang, D. Li, L. Xi, and E. Mylonakis, "Caenorhabditis elegans: A Simple Nematode Infection Model for Penicillium marneffei", PLoS ONE, vol. 9, pp. e108764, 2014. http://dx.doi.org/10.1371/journal.pone.0108764
- M.T. Glittenberg, S. Silas, D.M. MacCallum, N.A.R. Gow, and P. Ligoxygakis, "Wild-type Drosophila melanogaster as an alternative model system for investigating the pathogenicity of Candida albicans", Disease Models & Mechanisms, vol. 4, pp. 504-514, 2011. http://dx.doi.org/10.1242/dmm.006619
- R.A. Zanette, and D.P. Kontoyiannis, "Paradoxical effect to caspofungin inCandidaspecies does not confer survival advantage in aDrosophilamodel of candidiasis", Virulence, vol. 4, pp. 497-498, 2013. http://dx.doi.org/10.4161/viru.25523
- M.S. Lionakis, and D.P. Kontoyiannis, "Drosophila melanogaster as a Model Organism for Invasive Aspergillosis", Host-Fungus Interactions, pp. 455-468, 2012. http://dx.doi.org/10.1007/978-1-61779-539-8_32
- R.E. Lewis, R. Ben-Ami, L. Best, N. Albert, T.J. Walsh, and D.P. Kontoyiannis, "Tacrolimus Enhances the Potency of Posaconazole Against Rhizopus oryzae In Vitro and in an Experimental Model of Mucormycosis", Journal of Infectious Diseases, vol. 207, pp. 834-841, 2012. http://dx.doi.org/10.1093/infdis/jis767
- S. Dananjaya, W. Erandani, C. Kim, C. Nikapitiya, J. Lee, and M. De Zoysa, "Comparative study on antifungal activities of chitosan nanoparticles and chitosan silver nano composites against Fusarium oxysporum species complex", International Journal of Biological Macromolecules, vol. 105, pp. 478-488, 2017. http://dx.doi.org/10.1016/j.ijbiomac.2017.07.056
- S. Dananjaya, R. Udayangani, S.Y. Shin, M. Edussuriya, C. Nikapitiya, J. Lee, and M. De Zoysa, "In vitro and in vivo antifungal efficacy of plant based lawsone against Fusarium oxysporum species complex", Microbiological Research, vol. 201, pp. 21-29, 2017. http://dx.doi.org/10.1016/j.micres.2017.04.011
- H. Li, H. Gong, Y. Qi, J. Li, X. Ji, J. Sun, R. Tian, H. Bao, X. Song, Q. Chen, and G. Liu, "In vitro and in vivo antifungal activities and mechanism of heteropolytungstates against Candida species", Scientific Reports, vol. 7, 2017. http://dx.doi.org/10.1038/s41598-017-17239-8
- S. Dorsaz, A.T. Coste, and D. Sanglard, "Red-Shifted Firefly Luciferase Optimized for Candida albicans In vivo Bioluminescence Imaging", Frontiers in Microbiology, vol. 8, 2017. http://dx.doi.org/10.3389/fmicb.2017.01478
- A.J. Lepak, M. Zhao, B. VanScoy, P.G. Ambrose, and D.R. Andes, "Pharmacodynamics of a Long-Acting Echinocandin, CD101, in a Neutropenic Invasive-Candidiasis Murine Model Using an Extended-Interval Dosing Design", Antimicrobial Agents and Chemotherapy, vol. 62, 2018. http://dx.doi.org/10.1128/AAC.02154-17
- R. Li, L. Zhang, H. Zhang, Y. Yi, L. Wang, L. Chen, and L. Zhang, "Protective effect of a novel antifungal peptide derived from human chromogranin a on the immunity of mice infected with Candida krusei", Experimental and Therapeutic Medicine, vol. 13, pp. 2429-2434, 2017. http://dx.doi.org/10.3892/etm.2017.4290
- S.A. Wring, R. Randolph, S. Park, G. Abruzzo, Q. Chen, A. Flattery, G. Garrett, M. Peel, R. Outcalt, K. Powell, M. Trucksis, D. Angulo, and K. Borroto-Esoda, "Preclinical Pharmacokinetics and Pharmacodynamic Target of SCY-078, a First-in-Class Orally Active Antifungal Glucan Synthesis Inhibitor, in Murine Models of Disseminated Candidiasis", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.02068-16
- S.S.W. Wong, R.Y.T. Kao, K.Y. Yuen, Y. Wang, D. Yang, L.P. Samaranayake, and C.J. Seneviratne, "In Vitro and In Vivo Activity of a Novel Antifungal Small Molecule against Candida Infections", PLoS ONE, vol. 9, pp. e85836, 2014. http://dx.doi.org/10.1371/journal.pone.0085836
-
C. Paulussen, G. Boulet, T. Bosschaerts, P. Cos, A. Fortin, and L. Maes, "Efficacy of oleylphosphocholine (Ol
PC ) in vitro and in a mouse model of invasive aspergillosis", Mycoses, vol. 58, pp. 127-132, 2015. http://dx.doi.org/10.1111/myc.12286 - S.S. Rathore, M. Isravel, S. Vellaisamy, D.R. Chellappan, L. Cheepurupalli, T. Raman, and J. Ramakrishnan, "Exploration of Antifungal and Immunomodulatory Potentials of a Furanone Derivative to Rescue Disseminated Cryptococosis in Mice", Scientific Reports, vol. 7, 2017. http://dx.doi.org/10.1038/s41598-017-15500-8
- P. Thomson, L. López-Fernández, J. Guarro, and J. Capilla, "Virulence and antifungal therapy of murine disseminated infection by Rhodotorula mucilaginosa", Diagnostic Microbiology and Infectious Disease, vol. 89, pp. 47-51, 2017. http://dx.doi.org/10.1016/j.diagmicrobio.2017.06.005
- K. Ishida, R.A. Castro, J.J. Torrado, D.R. Serrano, L.P. Borba-Santos, L.P. Quintella, W. de Souza, S. Rozental, and L.M. Lopes-Bezerra, "Efficacy of a poly-aggregated formulation of amphotericin B in treating systemic sporotrichosis caused by Sporothrix brasiliensis", Medical Mycology, vol. 56, pp. 288-296, 2017. http://dx.doi.org/10.1093/mmy/myx040
- D. Seleem, B. Benso, J. Noguti, V. Pardi, and R.M. Murata, "In Vitro and In Vivo Antifungal Activity of Lichochalcone-A against Candida albicans Biofilms", PLOS ONE, vol. 11, pp. e0157188, 2016. http://dx.doi.org/10.1371/journal.pone.0157188
- T.J. Break, J.V. Desai, M. Natarajan, E.M.N. Ferre, C. Henderson, A.M. Zelazny, U. Siebenlist, W.J. Hoekstra, R.J. Schotzinger, E.P. Garvey, and M.S. Lionakis, "VT-1161 protects mice against oropharyngeal candidiasis caused by fluconazole-susceptible and -resistant Candida albicans", Journal of Antimicrobial Chemotherapy, vol. 73, pp. 151-155, 2017. http://dx.doi.org/10.1093/jac/dkx352
- T.V.M. Vila, A.K. Chaturvedi, S. Rozental, and J.L. Lopez-Ribot, "In Vitro Activity of Miltefosine against Candida albicans under Planktonic and Biofilm Growth Conditions and In Vivo Efficacy in a Murine Model of Oral Candidiasis", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 7611-7620, 2015. http://dx.doi.org/10.1128/AAC.01890-15
- K. Hata, T. Horii, M. Miyazaki, N. Watanabe, M. Okubo, J. Sonoda, K. Nakamoto, K. Tanaka, S. Shirotori, N. Murai, S. Inoue, M. Matsukura, S. Abe, K. Yoshimatsu, and M. Asada, "Efficacy of Oral E1210, a New Broad-Spectrum Antifungal with a Novel Mechanism of Action, in Murine Models of Candidiasis, Aspergillosis, and Fusariosis", Antimicrobial Agents and Chemotherapy, vol. 55, pp. 4543-4551, 2011. http://dx.doi.org/10.1128/AAC.00366-11
- A.H. Groll, and T.J. Walsh, "Antifungal efficacy and pharmacodynamics of posaconazole in experimental models of invasive fungal infections", Mycoses, vol. 49, pp. 7-16, 2006. http://dx.doi.org/10.1111/j.1439-0507.2006.01296.x
- D. Pietrella, B. Enjalbert, U. Zeidler, S. Znaidi, A. Rachini, A. Vecchiarelli, and C. d’Enfert, "A Luciferase Reporter for Gene Expression Studies and Dynamic Imaging of Superficial Candida albicans Infections", Host-Fungus Interactions, pp. 537-546, 2012. http://dx.doi.org/10.1007/978-1-61779-539-8_39
- T. Ci, L. Yuan, X. Bao, Y. Hou, H. Wu, H. Sun, D. Cao, and X. Ke, "Development and anti-Candida evaluation of the vaginal delivery system of amphotericin B nanosuspension-loaded thermogel", Journal of Drug Targeting, vol. 26, pp. 829-839, 2018. http://dx.doi.org/10.1080/1061186X.2018.1434660
- B.M. Peters, A. Luna-Tapia, H. Tournu, J.M. Rybak, P.D. Rogers, and G.E. Palmer, "An Azole-Tolerant Endosomal Trafficking Mutant of Candida albicans Is Susceptible to Azole Treatment in a Mouse Model of Vaginal Candidiasis", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.00084-17
- M. Gao, H. Wang, and L. Zhu, "Quercetin Assists Fluconazole to Inhibit Biofilm Formations of Fluconazole-Resistant Candida Albicans in In Vitro and In Vivo Antifungal Managements of Vulvovaginal Candidiasis", Cellular Physiology and Biochemistry, vol. 40, pp. 727-742, 2016. http://dx.doi.org/10.1159/000453134
- A. Bozó, M. Domán, L. Majoros, G. Kardos, I. Varga, and R. Kovács, "The in vitro and in vivo efficacy of fluconazole in combination with farnesol against Candida albicans isolates using a murine vulvovaginitis model", Journal of Microbiology, vol. 54, pp. 753-760, 2016. http://dx.doi.org/10.1007/s12275-016-6298-y
- D. Andes, J. Nett, P. Oschel, R. Albrecht, K. Marchillo, and A. Pitula, "Development and Characterization of an In Vivo Central Venous Catheter Candida albicans Biofilm Model", Infection and Immunity, vol. 72, pp. 6023-6031, 2004. http://dx.doi.org/10.1128/IAI.72.10.6023-6031.2004
- J.E. Nett, K. Marchillo, C.A. Spiegel, and D.R. Andes, "Development and Validation of an In Vivo Candida albicans Biofilm Denture Model", Infection and Immunity, vol. 78, pp. 3650-3659, 2010. http://dx.doi.org/10.1128/IAI.00480-10
-
G. Vande Velde, S. Kucharíková, S. Schrevens, U. Himmelreich, and P. Van Dijck, "Towards non‐invasive monitoring of pathogen–host interactions during
C andida albicans biofilm formation using in vivo bioluminescence", Cellular Microbiology, vol. 16, pp. 115-130, 2013. http://dx.doi.org/10.1111/cmi.12184 - H. Wu, S. Liu, N. Wiradharma, Z.Y. Ong, Y. Li, Y.Y. Yang, and J.Y. Ying, "Short Synthetic α‐Helical‐Forming Peptide Amphiphiles for Fungal Keratitis Treatment In Vivo", Advanced Healthcare Materials, vol. 6, 2017. http://dx.doi.org/10.1002/adhm.201600777
- A. Silva-Dias, I.M. Miranda, J. Branco, L. Cobrado, M. Monteiro-Soares, C. Pina-Vaz, and A.G. Rodrigues, "In vitro antifungal activity and in vivo antibiofilm activity of cerium nitrate against Candida species", Journal of Antimicrobial Chemotherapy, vol. 70, pp. 1083-1093, 2015. http://dx.doi.org/10.1093/jac/dku511
- A.L. Lazzell, A.K. Chaturvedi, C.G. Pierce, D. Prasad, P. Uppuluri, and J.L. Lopez-Ribot, "Treatment and prevention of Candida albicans biofilms with caspofungin in a novel central venous catheter murine model of candidiasis", Journal of Antimicrobial Chemotherapy, vol. 64, pp. 567-570, 2009. http://dx.doi.org/10.1093/jac/dkp242
- C.E. Negri, A. Johnson, L. McEntee, H. Box, S. Whalley, J.A. Schwartz, V. Ramos-Martín, J. Livermore, R. Kolamunnage-Dona, A.L. Colombo, and W.W. Hope, "Pharmacodynamics of the Novel Antifungal Agent F901318 for Acute Sinopulmonary Aspergillosis Caused by Aspergillus flavus", The Journal of Infectious Diseases, vol. 217, pp. 1118-1127, 2017. http://dx.doi.org/10.1093/infdis/jix479
- T. Colley, G. Sehra, A. Chowdhary, A. Alanio, S.L. Kelly, Y. Kizawa, D. Armstrong-James, M.C. Fisher, A.G.S. Warrilow, J.E. Parker, D.E. Kelly, G. Kimura, Y. Nishimoto, M. Sunose, S. Onions, D. Crepin, F. Lagasse, M. Crittall, J. Shannon, M. McConville, J. King-Underwood, A. Naylor, S. Bretagne, J. Murray, K. Ito, P. Strong, and G. Rapeport, "In Vitro and In Vivo Efficacy of a Novel and Long-Acting Fungicidal Azole, PC1244, on Aspergillus fumigatus Infection", Antimicrobial Agents and Chemotherapy, vol. 62, 2018. http://dx.doi.org/10.1128/AAC.01941-17
- G. Kimura, T. Nakaoki, T. Colley, G. Rapeport, P. Strong, K. Ito, and Y. Kizawa, "In Vivo Biomarker Analysis of the Effects of Intranasally Dosed PC945, a Novel Antifungal Triazole, on Aspergillus fumigatus Infection in Immunocompromised Mice", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.00124-17
- S. Seyedmousavi, J.W. Mouton, W.J.G. Melchers, and P.E. Verweij, "In Vivo Efficacy of Liposomal Amphotericin B against Wild-Type and Azole-Resistant Aspergillus fumigatus Isolates in Two Different Immunosuppression Models of Invasive Aspergillosis", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.02479-16
- H. Kai, M. Yamashita, I. Nakamura, K. Yoshikawa, K. Nitta, M. Watanabe, N. Inamura, and A. Fujie, "Synergistic antifungal activity of KB425796-C in combination with micafungin against Aspergillus fumigatus and its efficacy in murine infection models", The Journal of Antibiotics, vol. 66, pp. 479-484, 2013. http://dx.doi.org/10.1038/ja.2013.57
- G.L. Nixon, L. McEntee, A. Johnson, N. Farrington, S. Whalley, J. Livermore, C. Natal, G. Washbourn, J. Bibby, N. Berry, J. Lestner, M. Truong, A. Owen, D. Lalloo, I. Charles, and W. Hope, "Repurposing and Reformulation of the Antiparasitic Agent Flubendazole for Treatment of Cryptococcal Meningoencephalitis, a Neglected Fungal Disease", Antimicrobial Agents and Chemotherapy, vol. 62, 2018. http://dx.doi.org/10.1128/AAC.01909-17
- J.R.A. Santos, N.Q. Ribeiro, R.W. Bastos, R.A. Holanda, L.C. Silva, E.R. Queiroz, and D.A. Santos, "High-dose fluconazole in combination with amphotericin B is more efficient than monotherapy in murine model of cryptococcosis", Scientific Reports, vol. 7, 2017. http://dx.doi.org/10.1038/s41598-017-04588-7
- J.J. Jandú, M.C. Costa, J.R.A. Santos, F.M. Andrade, T.F. Magalhães, M.V. Silva, M.C.A.B. Castro, L.C.B.B. Coelho, A.G. Gomes, T.A. Paixão, D.A. Santos, and M.T.S. Correia, "Treatment with pCramoll Alone and in Combination with Fluconazole Provides Therapeutic Benefits in C. gattii Infected Mice", Frontiers in Cellular and Infection Microbiology, vol. 7, 2017. http://dx.doi.org/10.3389/fcimb.2017.00211
- H. Nishikawa, Y. Fukuda, J. Mitsuyama, M. Tashiro, A. Tanaka, T. Takazono, T. Saijo, K. Yamamoto, S. Nakamura, Y. Imamura, T. Miyazaki, H. Kakeya, Y. Yamamoto, K. Yanagihara, H. Mukae, S. Kohno, and K. Izumikawa, "In vitro and in vivo antifungal activities of T-2307, a novel arylamidine, against Cryptococcus gattii: an emerging fungal pathogen", Journal of Antimicrobial Chemotherapy, vol. 72, pp. 1709-1713, 2017. http://dx.doi.org/10.1093/jac/dkx020
- N.P. Wiederhold, L.F. Shubitz, L.K. Najvar, R. Jaramillo, M. Olivo, G. Catano, H.T. Trinh, C.M. Yates, R.J. Schotzinger, E.P. Garvey, and T.F. Patterson, "The Novel Fungal Cyp51 Inhibitor VT-1598 Is Efficacious in Experimental Models of Central Nervous System Coccidioidomycosis Caused by Coccidioides posadasii and Coccidioides immitis", Antimicrobial Agents and Chemotherapy, vol. 62, 2018. http://dx.doi.org/10.1128/AAC.02258-17
- B. López-García, P.H. Lee, K. Yamasaki, and R.L. Gallo, "Anti-Fungal Activity of Cathelicidins and their Potential Role in Candida albicans Skin Infection", Journal of Investigative Dermatology, vol. 125, pp. 108-115, 2005. http://dx.doi.org/10.1111/j.0022-202X.2005.23713.x
- R. Ben-Ami, R.E. Lewis, K. Leventakos, J. Latgé, and D.P. Kontoyiannis, "Cutaneous Model of Invasive Aspergillosis", Antimicrobial Agents and Chemotherapy, vol. 54, pp. 1848-1854, 2010. http://dx.doi.org/10.1128/AAC.01504-09
- S. Kucharíková, H. Tournu, M. Holtappels, P. Van Dijck, and K. Lagrou, "In Vivo Efficacy of Anidulafungin against Mature Candida albicans Biofilms in a Novel Rat Model of Catheter-Associated Candidiasis", Antimicrobial Agents and Chemotherapy, vol. 54, pp. 4474-4475, 2010. http://dx.doi.org/10.1128/AAC.00697-10
- A. Bink, S. Kucharíková, B. Neirinck, J. Vleugels, P. Van Dijck, B.P.A. Cammue, and K. Thevissen, "The Nonsteroidal Antiinflammatory Drug Diclofenac Potentiates the In Vivo Activity of Caspofungin Against Candida albicans Biofilms", The Journal of Infectious Diseases, vol. 206, pp. 1790-1797, 2012. http://dx.doi.org/10.1093/infdis/jis594
- S. Kucharíková, N. Sharma, I. Spriet, J. Maertens, P. Van Dijck, and K. Lagrou, "Activities of Systemically Administered Echinocandins against In Vivo Mature Candida albicans Biofilms Developed in a Rat Subcutaneous Model", Antimicrobial Agents and Chemotherapy, vol. 57, pp. 2365-2368, 2013. http://dx.doi.org/10.1128/AAC.02288-12
- S. Kucharíková, B. Neirinck, N. Sharma, J. Vleugels, K. Lagrou, and P. Van Dijck, "In vivo Candida glabrata biofilm development on foreign bodies in a rat subcutaneous model", Journal of Antimicrobial Chemotherapy, vol. 70, pp. 846-856, 2014. http://dx.doi.org/10.1093/jac/dku447
- M. Holtappels, E. Swinnen, L. De Groef, J. Wuyts, L. Moons, K. Lagrou, P. Van Dijck, and S. Kucharíková, "Antifungal Activity of Oleylphosphocholine on In Vitro and In Vivo Candida albicans Biofilms", Antimicrobial Agents and Chemotherapy, vol. 62, 2018. http://dx.doi.org/10.1128/AAC.01767-17
- D. Li, L. Zhao, E. Mylonakis, G. Hu, Y. Zou, T. Huang, L. Yan, Y. Wang, and Y. Jiang, "In Vitro and In Vivo Activities of Pterostilbene against Candida albicans Biofilms", Antimicrobial Agents and Chemotherapy, vol. 58, pp. 2344-2355, 2014. http://dx.doi.org/10.1128/AAC.01583-13
- E.M. Maiolo, A. Oliva, U. Furustrand Tafin, N. Perrotet, O. Borens, and A. Trampuz, "Antifungal activity against planktonic and biofilm Candida albicans in an experimental model of foreign-body infection", Journal of Infection, vol. 72, pp. 386-392, 2016. http://dx.doi.org/10.1016/j.jinf.2015.12.008
- J. Zhao, Y. Cheng, X. Song, C. Wang, G. Su, and Z. Liu, "A Comparative Treatment Study of Intravitreal Voriconazole and Liposomal Amphotericin B in anAspergillus fumigatusEndophthalmitis Model", Investigative Opthalmology & Visual Science, vol. 56, pp. 7369, 2015. http://dx.doi.org/10.1167/iovs.15-17266
- N.P. Wiederhold, L.K. Najvar, S. Matsumoto, R.A. Bocanegra, M.L. Herrera, B.L. Wickes, W.R. Kirkpatrick, and T.F. Patterson, "Efficacy of the Investigational Echinocandin ASP9726 in a Guinea Pig Model of Invasive Pulmonary Aspergillosis", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 2875-2881, 2015. http://dx.doi.org/10.1128/AAC.04857-14
- W.R. Kirkpatrick, L.K. Najvar, R. Bocanegra, T.F. Patterson, and J.R. Graybill, "New Guinea Pig Model of Cryptococcal Meningitis", Antimicrobial Agents and Chemotherapy, vol. 51, pp. 3011-3013, 2007. http://dx.doi.org/10.1128/AAC.00085-07
- E.P. Garvey, W.J. Hoekstra, R.J. Schotzinger, J.D. Sobel, E.A. Lilly, and P.L. Fidel, "Efficacy of the Clinical Agent VT-1161 against Fluconazole-Sensitive and -Resistant Candida albicans in a Murine Model of Vaginal Candidiasis", Antimicrobial Agents and Chemotherapy, vol. 59, pp. 5567-5573, 2015. http://dx.doi.org/10.1128/AAC.00185-15
- M. Dhandapani, and A. Goldman, "Preclinical Cancer Models and Biomarkers for Drug Development: New Technologies and Emerging Tools", Journal of Molecular Biomarkers & Diagnosis, vol. 08, 2017. http://dx.doi.org/10.4172/2155-9929.1000356
- B. Ho, N. Pek, and B. Soh, "Disease Modeling Using 3D Organoids Derived from Human Induced Pluripotent Stem Cells", International Journal of Molecular Sciences, vol. 19, pp. 936, 2018. http://dx.doi.org/10.3390/ijms19040936
- M. Crespo, E. Vilar, S. Tsai, K. Chang, S. Amin, T. Srinivasan, T. Zhang, N.H. Pipalia, H.J. Chen, M. Witherspoon, M. Gordillo, J.Z. Xiang, F.R. Maxfield, S. Lipkin, T. Evans, and S. Chen, "Colonic organoids derived from human induced pluripotent stem cells for modeling colorectal cancer and drug testing", Nature Medicine, vol. 23, pp. 878-884, 2017. http://dx.doi.org/10.1038/nm.4355
- A. Raz-Pasteur, Y. Ullmann, and I. Berdicevsky, "The Pathogenesis of Candida Infections in a Human Skin Model: Scanning Electron Microscope Observations", ISRN Dermatology, vol. 2011, pp. 1-6, 2011. http://dx.doi.org/10.5402/2011/150642
- L.J. Barkal, C.L. Procknow, Y.R. Álvarez-García, M. Niu, J.A. Jiménez-Torres, R.A. Brockman-Schneider, J.E. Gern, L.C. Denlinger, A.B. Theberge, N.P. Keller, E. Berthier, and D.J. Beebe, "Microbial volatile communication in human organotypic lung models", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/s41467-017-01985-4
- S. Brunke, J. Quintin, L. Kasper, I.D. Jacobsen, M.E. Richter, E. Hiller, T. Schwarzmüller, C. d'Enfert, K. Kuchler, S. Rupp, B. Hube, and D. Ferrandon, "Of mice, flies – and men? Comparing fungal infection models for large-scale screening efforts", Disease Models & Mechanisms, vol. 8, pp. 473-486, 2015. http://dx.doi.org/10.1242/dmm.019901
- W. Kim, G.L. Hendricks, K. Lee, and E. Mylonakis, "An update on the use ofC. elegansfor preclinical drug discovery: screening and identifying anti-infective drugs", Expert Opinion on Drug Discovery, vol. 12, pp. 625-633, 2017. http://dx.doi.org/10.1080/17460441.2017.1319358
- S. Limmer, J. Quintin, C. Hetru, and D. Ferrandon, "Virulence on the fly: Drosophila melanogaster as a model genetic organism to decipher host-pathogen interactions.", Current drug targets, 2011. http://www.ncbi.nlm.nih.gov/pubmed/21366519
- A.S. Gilbert, P.I. Seoane, P. Sephton-Clark, A. Bojarczuk, R. Hotham, E. Giurisato, A.R. Sarhan, A. Hillen, G.V. Velde, N.S. Gray, D.R. Alessi, D.L. Cunningham, C. Tournier, S.A. Johnston, and R.C. May, "Vomocytosis of live pathogens from macrophages is regulated by the atypical MAP kinase ERK5", Science Advances, vol. 3, 2017. http://dx.doi.org/10.1126/sciadv.1700898
- Y. Kochi, Y. Matsumoto, K. Sekimizu, and C. Kaito, "Two-spotted cricket as an animal infection model of human pathogenic fungi.", Drug discoveries & therapeutics, 2017. http://www.ncbi.nlm.nih.gov/pubmed/29081438
- U. Binder, E. Maurer, and C. Lass-Flörl, "Galleria mellonella: An invertebrate model to study pathogenicity in correctly defined fungal species", Fungal Biology, vol. 120, pp. 288-295, 2016. http://dx.doi.org/10.1016/j.funbio.2015.06.002
- S. Krappmann, "Lightning up the worm: How to probe fungal virulence in an alternative mini-host by bioluminescence", Virulence, vol. 6, pp. 727-729, 2015. http://dx.doi.org/10.1080/21505594.2015.1103428
- C. Paulussen, G.A. Boulet, P. Cos, P. Delputte, and L.J. Maes, "Animal models of invasive aspergillosis for drug discovery", Drug Discovery Today, vol. 19, pp. 1380-1386, 2014. http://dx.doi.org/10.1016/j.drudis.2014.06.006
- R.E. Lewis, and P.E. Verweij, "Animal Models for Studying Triazole Resistance in Aspergillus fumigatus", The Journal of Infectious Diseases, vol. 216, pp. S466-S473, 2017. http://dx.doi.org/10.1093/infdis/jix222
- S.M. Saporito-Irwin, C.E. Birse, P.S. Sypherd, and W.A. Fonzi, "PHR1, a pH-regulated gene of Candida albicans, is required for morphogenesis.", Molecular and cellular biology, 1995. http://www.ncbi.nlm.nih.gov/pubmed/7823929
- F.A. Mühlschlegel, and W.A. Fonzi, "PHR2 of Candida albicans encodes a functional homolog of the pH-regulated gene PHR1 with an inverted pattern of pH-dependent expression.", Molecular and cellular biology, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9315654
- F. De Bernardis, F.A. Mühlschlegel, A. Cassone, and W.A. Fonzi, "The pH of the host niche controls gene expression in and virulence of Candida albicans.", Infection and immunity, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9632601
- C. Popp, I.A.I. Hampe, T. Hertlein, K. Ohlsen, P.D. Rogers, and J. Morschhäuser, "Competitive Fitness of Fluconazole-Resistant Clinical Candida albicans Strains", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.00584-17
- E. Segal, and M. Frenkel, "Experimental In Vivo Models of Candidiasis", Journal of Fungi, vol. 4, pp. 21, 2018. http://dx.doi.org/10.3390/jof4010021
- H.A. Mohamed, R.R. Radwan, A.I. Raafat, and A.E. Ali, "Antifungal activity of oral (Tragacanth/acrylic acid) Amphotericin B carrier for systemic candidiasis: in vitro and in vivo study", Drug Delivery and Translational Research, vol. 8, pp. 191-203, 2017. http://dx.doi.org/10.1007/s13346-017-0452-x
- R. Semis, S.S. Nili, A. Munitz, Z. Zaslavsky, I. Polacheck, and E. Segal, "Pharmacokinetics, tissue distribution and immunomodulatory effect of intralipid formulation of nystatin in mice", Journal of Antimicrobial Chemotherapy, vol. 67, pp. 1716-1721, 2012. http://dx.doi.org/10.1093/jac/dks117
- D.M. Saunte, J.P. Hasselby, A. Brillowska-Dabrowska, N. Frimodt-Møller, E.L. Svejgaard, D. Linnemann, S.S. Nielsen, M. Hædersdal, and M.C. Arendrup, "Experimental guinea pig model of dermatophytosis: a simple and useful tool for the evaluation of new diagnostics and antifungals", Medical Mycology, vol. 46, pp. 303-313, 2008. http://dx.doi.org/10.1080/13693780801891732
- L. Cambier, M. Heinen, and B. Mignon, "Relevant Animal Models in Dermatophyte Research", Mycopathologia, vol. 182, pp. 229-240, 2016. http://dx.doi.org/10.1007/s11046-016-0079-3
- K. Maebashi, T. Itoyama, K. Uchida, N. Suegara, and H. Yamaguchi, "A novel model of cutaneous candidiasis produced in prednisolone-treated guinea-pigs.", Journal of medical and veterinary mycology : bi-monthly publication of the International Society for Human and Animal Mycology, 1994. http://www.ncbi.nlm.nih.gov/pubmed/7844701
- Z. Rhew, and Y. Han, "Synergic effect of combination of glycyrol and fluconazole against experimental cutaneous candidiasis due to Candida albicans", Archives of Pharmacal Research, vol. 39, pp. 1482-1489, 2016. http://dx.doi.org/10.1007/s12272-016-0824-7
- R. Ben-Ami, and D.P. Kontoyiannis, "A Nonlethal Murine Cutaneous Model of Invasive Aspergillosis", Host-Fungus Interactions, pp. 569-582, 2012. http://dx.doi.org/10.1007/978-1-61779-539-8_42
- A.F. BONK, L. FRIEDMAN, and V.J. DERBES, "Experimental dermatophytosis.", The Journal of investigative dermatology, 1962. http://www.ncbi.nlm.nih.gov/pubmed/13968812
- A. Treiber, W. Pittermann, and H. Schuppe, "Efficacy testing of antimycotic prophylactics in an animal model", International Journal of Hygiene and Environmental Health, vol. 204, pp. 239-243, 2001. http://dx.doi.org/10.1078/S1438-4639(04)70100-5
- J.H. Greenberg, R.D. King, S. Krebs, and R. Field, "A quantitative dermatophyte infection model in the guinea pig--a parallel to the quantitated human infection model.", The Journal of investigative dermatology, 1976. http://www.ncbi.nlm.nih.gov/pubmed/1003008
- A. Polak, "Experimental models in antifungal chemotherapy.", Mycoses, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9610129
- D.M. Saunte, F. Simmel, N. Frimodt-Moller, L.B. Stolle, E.L. Svejgaard, M. Haedersdal, C. Kloft, and M.C. Arendrup, "In Vivo Efficacy and Pharmacokinetics of Voriconazole in an Animal Model of Dermatophytosis", Antimicrobial Agents and Chemotherapy, vol. 51, pp. 3317-3321, 2007. http://dx.doi.org/10.1128/AAC.01185-06
- K. de Wit, C. Paulussen, A. Matheeussen, K. van Rossem, P. Cos, and L. Maes, "In Vitro Profiling of Pramiconazole and In Vivo Evaluation in Microsporum canis Dermatitis and Candida albicans Vaginitis Laboratory Models", Antimicrobial Agents and Chemotherapy, vol. 54, pp. 4927-4929, 2010. http://dx.doi.org/10.1128/AAC.00730-10
- T. Shimamura, N. Kubota, S. Nagasaka, T. Suzuki, H. Mukai, and K. Shibuya, "Establishment of a Novel Model of Onychomycosis in Rabbits for Evaluation of Antifungal Agents", Antimicrobial Agents and Chemotherapy, vol. 55, pp. 3150-3155, 2011. http://dx.doi.org/10.1128/AAC.00399-11
- Y. Tatsumi, M. Yokoo, H. Senda, and K. Kakehi, "Therapeutic efficacy of topically applied KP-103 against experimental tinea unguium in guinea pigs in comparison with amorolfine and terbinafine.", Antimicrobial agents and chemotherapy, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12435679
- J.R. Naglik, P.L. Fidel, and F.C. Odds, "Animal models of mucosal Candida infection", FEMS Microbiology Letters, vol. 283, pp. 129-139, 2008. http://dx.doi.org/10.1111/j.1574-6968.2008.01160.x
- J. Yano, and P.L. Fidel, Jr., "Protocols for Vaginal Inoculation and Sample Collection in the Experimental Mouse Model of <em>Candida</em> vaginitis", Journal of Visualized Experiments, 2011. http://dx.doi.org/10.3791/3382
- E.E. Nash, B.M. Peters, E.A. Lilly, M.C. Noverr, and P.L. Fidel, "A Murine Model of Candida glabrata Vaginitis Shows No Evidence of an Inflammatory Immunopathogenic Response", PLOS ONE, vol. 11, pp. e0147969, 2016. http://dx.doi.org/10.1371/journal.pone.0147969
- E.F. Kong, S. Kucharíková, P. Van Dijck, B.M. Peters, M.E. Shirtliff, and M.A. Jabra-Rizk, "Clinical Implications of Oral Candidiasis: Host Tissue Damage and Disseminated Bacterial Disease", Infection and Immunity, vol. 83, pp. 604-613, 2015. http://dx.doi.org/10.1128/IAI.02843-14
- L. Wang, C. Wang, H. Mei, Y. Shen, G. Lv, R. Zeng, P. Zhan, D. Li, and W. Liu, "Combination of Estrogen and Immunosuppressive Agents to Establish a Mouse Model of Candidiasis with Concurrent Oral and Vaginal Mucosal Infection", Mycopathologia, vol. 181, pp. 29-39, 2015. http://dx.doi.org/10.1007/s11046-015-9947-5
- L.R. Hanson, J.M. Fine, A.L. Svitak, and K.A. Faltesek, "Intranasal Administration of CNS Therapeutics to Awake Mice", Journal of Visualized Experiments, 2013. http://dx.doi.org/10.3791/4440
- R.A. Larsen, M. Bauer, A.M. Thomas, and J.R. Graybill, "Amphotericin B and fluconazole, a potent combination therapy for cryptococcal meningitis.", Antimicrobial agents and chemotherapy, 2004. http://www.ncbi.nlm.nih.gov/pubmed/14982793
- F. Dromer, C. Bernede-Bauduin, D. Guillemot, O. Lortholary, and . , "Major Role for Amphotericin B–Flucytosine Combination in Severe Cryptococcosis", PLoS ONE, vol. 3, pp. e2870, 2008. http://dx.doi.org/10.1371/journal.pone.0002870
- A. Ullmann, J. Aguado, S. Arikan-Akdagli, D. Denning, A. Groll, K. Lagrou, C. Lass-Flörl, R. Lewis, P. Munoz, P. Verweij, A. Warris, F. Ader, M. Akova, M. Arendrup, R. Barnes, C. Beigelman-Aubry, S. Blot, E. Bouza, R. Brüggemann, D. Buchheidt, J. Cadranel, E. Castagnola, A. Chakrabarti, M. Cuenca-Estrella, G. Dimopoulos, J. Fortun, J. Gangneux, J. Garbino, W. Heinz, R. Herbrecht, C. Heussel, C. Kibbler, N. Klimko, B. Kullberg, C. Lange, T. Lehrnbecher, J. Löffler, O. Lortholary, J. Maertens, O. Marchetti, J. Meis, L. Pagano, P. Ribaud, M. Richardson, E. Roilides, M. Ruhnke, M. Sanguinetti, D. Sheppard, J. Sinkó, A. Skiada, M. Vehreschild, C. Viscoli, and O. Cornely, "Diagnosis and management of Aspergillus diseases: executive summary of the 2017 ESCMID-ECMM-ERS guideline", Clinical Microbiology and Infection, vol. 24, pp. e1-e38, 2018. http://dx.doi.org/10.1016/j.cmi.2018.01.002
- X. Zhou, Y. Dang, G. Wang, and X. Jin, "Effects ofAspergillus fumigatuson glucocorticoid receptor and β2-adrenergic receptor expression in a rat model of asthma", Experimental Lung Research, vol. 43, pp. 197-207, 2017. http://dx.doi.org/10.1080/01902148.2017.1339142
- K. Schäfer, A. Di Pietro, N.A.R. Gow, and D. MacCallum, "Murine Model for Fusarium oxysporum Invasive Fusariosis Reveals Organ-Specific Structures for Dissemination and Long-Term Persistence", PLoS ONE, vol. 9, pp. e89920, 2014. http://dx.doi.org/10.1371/journal.pone.0089920
- F. Fernández-Silva, J. Capilla, E. Mayayo, and J. Guarro, "Efficacy of Posaconazole in Murine Experimental Sporotrichosis", Antimicrobial Agents and Chemotherapy, vol. 56, pp. 2273-2277, 2012. http://dx.doi.org/10.1128/AAC.05376-11
- V. Petraitis, R. Petraitiene, W.W. Hope, and T.J. Walsh, "Endpoint Assessment in Rabbit Models of Invasive Pulmonary Aspergillosis and Mucormycosis", Methods in Molecular Biology, pp. 259-277, 2017. http://dx.doi.org/10.1007/978-1-4939-7104-6_18
- T. Gebremariam, S. Alkhazraji, C. Baldin, L. Kovanda, N.P. Wiederhold, and A.S. Ibrahim, "Prophylaxis with Isavuconazole or Posaconazole Protects Immunosuppressed Mice from Pulmonary Mucormycosis", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.02589-16
- B. Lelièvre, P. Legras, C. Godon, F. Franconi, J. Saint-André, J. Bouchara, and B. Diquet, "Experimental Models of Disseminated Scedosporiosis with Cerebral Involvement", Journal of Pharmacology and Experimental Therapeutics, vol. 345, pp. 198-205, 2013. http://dx.doi.org/10.1124/jpet.112.201541
- D. Sheppard, K. Marr, D. Fredricks, L. Chiang, T. Doedt, and S. Filler, "Comparison of three methodologies for the determination of pulmonary fungal burden in experimental murine aspergillosis", Clinical Microbiology and Infection, vol. 12, pp. 376-380, 2006. http://dx.doi.org/10.1111/j.1469-0691.2005.01349.x
- D. Prieto, E. Román, I. Correia, and J. Pla, "The HOG Pathway Is Critical for the Colonization of the Mouse Gastrointestinal Tract by Candida albicans", PLoS ONE, vol. 9, pp. e87128, 2014. http://dx.doi.org/10.1371/journal.pone.0087128
- H. Sandovsky-Losica, L. Barr-Nea, and E. Segal, "Fatal systemic candidiasis of gastrointestinal origin: an experimental model in mice compromised by anti-cancer treatment.", Journal of medical and veterinary mycology : bi-monthly publication of the International Society for Human and Animal Mycology, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1517959
- M. Kobayashi-Sakamoto, R. Tamai, E. Isogai, and Y. Kiyoura, "Gastrointestinal colonisation and systemic spread of Candida albicans in mice treated with antibiotics and prednisolone", Microbial Pathogenesis, vol. 117, pp. 191-199, 2018. http://dx.doi.org/10.1016/j.micpath.2018.02.043
- D. Kadosh, L.K. Najvar, R. Bocanegra, M. Olivo, W.R. Kirkpatrick, N.P. Wiederhold, and T.F. Patterson, "Effect of Antifungal Treatment in a Diet-Based Murine Model of Disseminated Candidiasis Acquired via the Gastrointestinal Tract", Antimicrobial Agents and Chemotherapy, vol. 60, pp. 6703-6708, 2016. http://dx.doi.org/10.1128/AAC.01144-16
- K.R. Healey, Y. Nagasaki, M. Zimmerman, M. Kordalewska, S. Park, Y. Zhao, and D.S. Perlin, "The Gastrointestinal Tract Is a Major Source of Echinocandin Drug Resistance in a Murine Model of Candida glabrata Colonization and Systemic Dissemination", Antimicrobial Agents and Chemotherapy, vol. 61, 2017. http://dx.doi.org/10.1128/AAC.01412-17
- J.L. Hoeflinger, D.A. Coleman, S. Oh, M.J. Miller, and L.L. Hoyer, "A piglet model for studyingCandida albicanscolonization of the human oro-gastrointestinal tract", FEMS Microbiology Letters, vol. 357, pp. 10-15, 2014. http://dx.doi.org/10.1111/1574-6968.12500
- D.M. MacCallum, and F.C. Odds, "Temporal events in the intravenous challenge model for experimental Candida albicans infections in female mice", Mycoses, vol. 48, pp. 151-161, 2005. http://dx.doi.org/10.1111/j.1439-0507.2005.01121.x
- D.M. MacCallum, "Mouse Model of Invasive Fungal Infection", Mouse Models of Innate Immunity, pp. 145-153, 2013. http://dx.doi.org/10.1007/978-1-62703-481-4_17
- Y. Wu, F. Min, J. Pan, J. Wang, W. Yuan, Y. Zhang, R. Huang, and L. Zhang, "SystemicCandida parapsilosisInfection Model in Immunosuppressed ICR Mice and Assessing the Antifungal Efficiency of Fluconazole", Veterinary Medicine International, vol. 2015, pp. 1-7, 2015. http://dx.doi.org/10.1155/2015/370641
- F.C. Odds, L. Van Nuffel, and N.A.R. Gow, "Survival in experimental Candida albicans infections depends on inoculum growth conditions as well as animal host", Microbiology, vol. 146, pp. 1881-1889, 2000. http://dx.doi.org/10.1099/00221287-146-8-1881
-
V. Salas, F.J. Pastor, E. Calvo, E. Mayayo, G. Quindós, A.J. Carrillo, and J. Guarro, "Anidulafungin in Treatment of Experimental Invasive Infection by Candida parapsilosis:
In Vitro
Activity, (1→3)-β-
d -Glucan and Mannan Serum Levels, Histopathological Findings, and In Vivo Efficacy", Antimicrobial Agents and Chemotherapy, vol. 55, pp. 4985-4989, 2011. http://dx.doi.org/10.1128/AAC.00500-11 - A.A. Sanchez, D.A. Johnston, C. Myers, J.E. Edwards, A.P. Mitchell, and S.G. Filler, "Relationship between Candida albicans virulence during experimental hematogenously disseminated infection and endothelial cell damage in vitro.", Infection and immunity, 2004. http://www.ncbi.nlm.nih.gov/pubmed/14688143
- H. Hänel, W. Raether, M. Uphoff, B. Braun, J. Kalisch, and S. Pastowsky, "A modified method for experimental candidosis in mice avoiding lethality.", Mycoses, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1435851
- R. Hajdu, R. Thompson, J.G. Sundelof, B.A. Pelak, F.A. Bouffard, J.F. Dropinski, and H. Kropp, "Preliminary animal pharmacokinetics of the parenteral antifungal agent MK-0991 (L-743,872).", Antimicrobial agents and chemotherapy, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9371330
- B. Damle, M. Stogniew, and J. Dowell, "Pharmacokinetics and Tissue Distribution of Anidulafungin in Rats", Antimicrobial Agents and Chemotherapy, vol. 52, pp. 2673-2676, 2008. http://dx.doi.org/10.1128/AAC.01596-07
- T. Niwa, Y. Yokota, A. Tokunaga, Y. Yamato, A. Kagayama, T. Fujiwara, J. Hatakeyama, M. Anezaki, Y. Ohtsuka, and A. Takagi, "Tissue distribution after intravenous dosing of micafungin, an antifungal drug, to rats.", Biological & pharmaceutical bulletin, 2004. http://www.ncbi.nlm.nih.gov/pubmed/15256761
- U. Theuretzbacher, "Pharmacokinetics/pharmacodynamics of echinocandins", European Journal of Clinical Microbiology & Infectious Diseases, vol. 23, pp. 805-812, 2004. http://dx.doi.org/10.1007/s10096-004-1228-z
- S.J. Roffey, S. Cole, P. Comby, D. Gibson, S.G. Jezequel, A.N.R. Nedderman, D.A. Smith, D.K. Walker, and N. Wood, "The disposition of voriconazole in mouse, rat, rabbit, guinea pig, dog, and human.", Drug metabolism and disposition: the biological fate of chemicals, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12756205
- G.V. Velde, S. Kucharíková, P. Van Dijck, and U. Himmelreich, "Bioluminescence Imaging of Fungal Biofilm Development in Live Animals", Methods in Molecular Biology, pp. 153-167, 2013. http://dx.doi.org/10.1007/978-1-62703-718-1_13
- B. Enjalbert, A. Rachini, G. Vediyappan, D. Pietrella, R. Spaccapelo, A. Vecchiarelli, A.J.P. Brown, and C. d'Enfert, "A Multifunctional, Synthetic Gaussia princeps Luciferase Reporter for Live Imaging of Candida albicans Infections", Infection and Immunity, vol. 77, pp. 4847-4858, 2009. http://dx.doi.org/10.1128/IAI.00223-09
- P. Mosci, E. Pericolini, E. Gabrielli, S. Kenno, S. Perito, F. Bistoni, C. d’Enfert, and A. Vecchiarelli, "A novel bioluminescence mouse model for monitoring oropharyngeal candidiasis in mice", Virulence, vol. 4, pp. 250-254, 2013. http://dx.doi.org/10.4161/viru.23529
- S. Kucharíková, G. Vande Velde, U. Himmelreich, and P. Van Dijck, "<em>Candida albicans</em> Biofilm Development on Medically-relevant Foreign Bodies in a Mouse Subcutaneous Model Followed by Bioluminescence Imaging", Journal of Visualized Experiments, 2015. http://dx.doi.org/10.3791/52239
- I.D. Jacobsen, A. Lüttich, O. Kurzai, B. Hube, and M. Brock, "In vivo imaging of disseminated murine Candida albicans infection reveals unexpected host sites of fungal persistence during antifungal therapy", Journal of Antimicrobial Chemotherapy, vol. 69, pp. 2785-2796, 2014. http://dx.doi.org/10.1093/jac/dku198
- L. Vanherp, J. Poelmans, A. Hillen, K. Govaerts, S. Belderbos, T. Buelens, K. Lagrou, U. Himmelreich, and G. Vande Velde, "Bronchoscopic fibered confocal fluorescence microscopy for longitudinal in vivo assessment of pulmonary fungal infections in free-breathing mice", Scientific Reports, vol. 8, 2018. http://dx.doi.org/10.1038/s41598-018-20545-4
- J. Poelmans, A. Hillen, L. Vanherp, K. Govaerts, J. Maertens, T. Dresselaers, U. Himmelreich, K. Lagrou, and G. Vande Velde, "Longitudinal, in vivo assessment of invasive pulmonary aspergillosis in mice by computed tomography and magnetic resonance imaging", Laboratory Investigation, vol. 96, pp. 692-704, 2016. http://dx.doi.org/10.1038/labinvest.2016.45
- M.H. Lee, G. Wiedman, S. Park, A. Mustaev, Y. Zhao, and D.S. Perlin, "A novel, tomographic imaging probe for rapid diagnosis of fungal keratitis", Medical Mycology, vol. 56, pp. 796-802, 2017. http://dx.doi.org/10.1093/mmy/myx125
- M. Ghannoum, E. Roilides, A. Katragkou, V. Petraitis, and T.J. Walsh, "The Role of Echinocandins in Candida Biofilm–Related Vascular Catheter Infections: In Vitro and In Vivo Model Systems", Clinical Infectious Diseases, vol. 61, pp. S618-S621, 2015. http://dx.doi.org/10.1093/cid/civ815
- J.E. Nett, and D. R Andes, "Fungal Biofilms: In Vivo Models for Discovery of Anti-Biofilm Drugs.", Microbiology spectrum, 2015. http://www.ncbi.nlm.nih.gov/pubmed/26397003
- M.K. Schinabeck, L.A. Long, M.A. Hossain, J. Chandra, P.K. Mukherjee, S. Mohamed, and M.A. Ghannoum, "Rabbit model of Candida albicans biofilm infection: liposomal amphotericin B antifungal lock therapy.", Antimicrobial agents and chemotherapy, 2004. http://www.ncbi.nlm.nih.gov/pubmed/15105127
- J. Chandra, L. Long, M.A. Ghannoum, and P.K. Mukherjee, "A rabbit model for evaluation of catheter-associated fungal biofilms", Virulence, vol. 2, pp. 466-474, 2011. http://dx.doi.org/10.4161/viru.2.5.16341
- H.T. Taff, K. Marchillo, and D.R. Andes, "Preparation of Candida albicans Biofilms Using an in vivo Rat Central Venous Catheter Model.", Bio-protocol, 2013. http://www.ncbi.nlm.nih.gov/pubmed/25927075
- P. Uppuluri, J. Nett, J. Heitman, and D. Andes, "Synergistic Effect of Calcineurin Inhibitors and Fluconazole against Candida albicans Biofilms", Antimicrobial Agents and Chemotherapy, vol. 52, pp. 1127-1132, 2008. http://dx.doi.org/10.1128/AAC.01397-07
- M. Řičicová, S. Kucharíková, H. Tournu, J. Hendrix, H. Bujdáková, J. Van Eldere, K. Lagrou, and P. Van Dijck, "Candida albicans biofilm formation in a new in vivo rat model", Microbiology, vol. 156, pp. 909-919, 2010. http://dx.doi.org/10.1099/mic.0.033530-0
- C.C. Johnson, A. Yu, H. Lee, P.L. Fidel, and M.C. Noverr, "Development of a Contemporary Animal Model of Candida albicans-Associated Denture Stomatitis Using a Novel Intraoral Denture System", Infection and Immunity, vol. 80, pp. 1736-1743, 2012. http://dx.doi.org/10.1128/IAI.00019-12
- T. Wu, W. Hu, L. Guo, M. Finnegan, D.J. Bradshaw, P. Webster, Z.G. Loewy, X. Zhou, W. Shi, and R. Lux, "Development of a New Model System to Study Microbial Colonization on Dentures", Journal of Prosthodontics, vol. 22, pp. 344-350, 2013. http://dx.doi.org/10.1111/jopr.12002
- R.D. Rossoni, P.P. de Barros, J.A. de Alvarenga, F.D.C. Ribeiro, M.D.S. Velloso, B.B. Fuchs, E. Mylonakis, A.O.C. Jorge, and J.C. Junqueira, "Antifungal activity of clinical Lactobacillus strains against Candida albicans biofilms: identification of potential probiotic candidates to prevent oral candidiasis", Biofouling, vol. 34, pp. 212-225, 2018. http://dx.doi.org/10.1080/08927014.2018.1425402
- R.D. Rossoni, M. dos Santos Velloso, L.M.A. Figueiredo, C.P. Martins, A.O.C. Jorge, and J.C. Junqueira, "Clinical strains of Lactobacillus reduce the filamentation of Candida albicans and protect Galleria mellonella against experimental candidiasis", Folia Microbiologica, vol. 63, pp. 307-314, 2017. http://dx.doi.org/10.1007/s12223-017-0569-9
- R.D. Rossoni, B.B. Fuchs, P.P. de Barros, M.D.S. Velloso, A.O.C. Jorge, J.C. Junqueira, and E. Mylonakis, "Lactobacillus paracasei modulates the immune system of Galleria mellonella and protects against Candida albicans infection", PLOS ONE, vol. 12, pp. e0173332, 2017. http://dx.doi.org/10.1371/journal.pone.0173332
- P. Albuquerque, and A. Casadevall, "Quorum sensing in fungi – a review", Medical Mycology, vol. 50, pp. 337-345, 2012. http://dx.doi.org/10.3109/13693786.2011.652201
- M. Cornet, and C. Gaillardin, "pH Signaling in Human Fungal Pathogens: a New Target for Antifungal Strategies", Eukaryotic Cell, vol. 13, pp. 342-352, 2014. http://dx.doi.org/10.1128/EC.00313-13
- F. Gerwien, V. Skrahina, L. Kasper, B. Hube, and S. Brunke, "Metals in fungal virulence", FEMS Microbiology Reviews, vol. 42, 2017. http://dx.doi.org/10.1093/femsre/fux050
- K.D. Gank, M.R. Yeaman, S. Kojima, N.Y. Yount, H. Park, J.E. Edwards, S.G. Filler, and Y. Fu, "SSD1 Is Integral to Host Defense Peptide Resistance in Candida albicans", Eukaryotic Cell, vol. 7, pp. 1318-1327, 2008. http://dx.doi.org/10.1128/EC.00402-07
- S. Jung, J.S. Finkel, N.V. Solis, S. Chaili, A.P. Mitchell, M.R. Yeaman, and S.G. Filler, "Bcr1 Functions Downstream of Ssd1 To Mediate Antimicrobial Peptide Resistance in Candida albicans", Eukaryotic Cell, vol. 12, pp. 411-419, 2013. http://dx.doi.org/10.1128/EC.00285-12
- S. Kucharíková, E. Gerits, K. De Brucker, A. Braem, K. Ceh, G. Majdič, T. Španič, E. Pogorevc, N. Verstraeten, H. Tournu, N. Delattin, F. Impellizzeri, M. Erdtmann, A. Krona, M. Lövenklev, M. Knezevic, M. Fröhlich, J. Vleugels, M. Fauvart, W.J. de Silva, K. Vandamme, J. Garcia-Forgas, B.P.A. Cammue, J. Michiels, P. Van Dijck, and K. Thevissen, "Covalent immobilization of antimicrobial agents on titanium preventsStaphylococcus aureusandCandida albicanscolonization and biofilm formation", Journal of Antimicrobial Chemotherapy, vol. 71, pp. 936-945, 2015. http://dx.doi.org/10.1093/jac/dkv437
ACKNOWLEDGMENTS
KT acknowledges receipt of a mandate of Industrial Research Fund (IOFm/05/022). JB acknowledges funding from the European Research Council Advanced Award 3400867/RAPLODAPT and the Israel Science Foundation grant # 314/13 (www.isf.il). NG acknowledges the Wellcome Trust and MRC for funding. CD acknowledges funding from the Agence Nationale de Recherche (ANR-10-LABX-62-IBEID). CJN acknowledges funding from the National Institutes of Health R35GM124594 and R21AI125801. AW is supported by the Wellcome Trust Strategic Award (grant 097377), the MRC Centre for Medical Mycology (grant MR/N006364/1) at the University of Aberdeen. MaCA: outside this study MaCA has received personal speaker’s honoraria the past five years from Astellas, Basilea, Gilead, MSD, Pfizer, T2Candida, and Novartis. She has received research grants and contract work paid to the Statens Serum Institute from Astellas, Basilea, Gilead, MSD, NovaBiotics, Pfizer, T2Biosystems, F2G, Cidara, and Amplyx. CAM acknowledges the Wellcome Trust and the MRC MR/N006364/1. PVD, TC and KT acknowledge the FWO research community: Biology and ecology of bacterial and fungal biofilms in humans (FWO WO.009.16N). AAB acknowledges the Deutsche Forschungsgemeinschaft – CRC FungiNet.
COPYRIGHT
© 2018

Methodologies for in vitro and in vivo evaluation of efficacy of antifungal and antibiofilm agents and surface coatings against fungal biofilms by Van Dijck et al. is licensed under a Creative Commons Attribution 4.0 International License.








