
Reviews:
Microbial Cell, Vol. 5, No. 12, pp. 525 - 544; doi: 10.15698/mic2018.12.659
Pathways of host cell exit by intracellular pathogens

1 Division of Enteropathogenic Bacteria and Legionella, Robert Koch Institute, Wernigerode, Germany.
2 Center for Infectious Diseases, University of Heidelberg, Germany.
3 Institute of Medical Microbiology and Hygiene, Medical Center – University of Freiburg, Germany.
4 Institute of Medical Microbiology, RWTH Aachen University Hospital, Germany.
5 Division of Cellular and Applied Infection Biology, Institute of Biology II, RWTH Aachen University, Germany.
# Authors listed in alphabetic order.
Keywords: host cell, egress, exit, pathogen, vacuolar escape, compartment, programmed cell death, membrane lysis.
Abbreviations:
ABA – abscisic acid,
ActA – actin assembly-inducing protein,
Arp2/3 – actin-related protein 2/3,
CPAF – chlamydial protease-like activity factor,
N-WASP – neuronal WASP,
PCD – programmed cell death.
PI – phosphatidylinositol,
PK – protein kinase,
PV - parasitophorous vacuole,
PVM – PV membrane,
RBC - red blood cell,
RBCM – RBC membrane,
WASP – Wiscott-Aldrich syndrome protein.
Received originally: 02/07/2018 Received in revised form: 10/09/2018
Accepted: 13/10/2018
Published: 18/10/2018
Correspondence:
Gabriele Pradel, Division of Cellular and Applied Infection Biology, RWTH Aachen University, Worringerweg 1, 52074 Aachen; phone: +49-(0)241-8020848; pradel@bio2.rwth-aachen.de
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Antje Flieger, Freddy Frischknecht, Georg Häcker, Mathias W. Hornef, Gabriele Pradel (2018). Pathways of host cell exit by intracellular pathogens. Microbial Cell 5(12): 525-544. doi: 10.15698/mic2018.12.659
Abstract
Host cell exit is a critical step in the life-cycle of intracellular pathogens, intimately linked to barrier penetration, tissue dissemination, inflammation, and pathogen transmission. Like cell invasion and intracellular survival, host cell exit represents a well-regulated program that has evolved during host-pathogen co-evolution and that relies on the dynamic and intricate interplay between multiple host and microbial factors. Three distinct pathways of host cell exit have been identified that are employed by three different taxa of intracellular pathogens, bacteria, fungi and protozoa, namely (i) the initiation of programmed cell death, (ii) the active breaching of host cell-derived membranes, and (iii) the induced membrane-dependent exit without host cell lysis. Strikingly, an increasing number of studies show that the majority of intracellular pathogens utilize more than one of these strategies, dependent on life-cycle stage, environmental factors and/or host cell type. This review summarizes the diverse exit strategies of intracellular-living bacterial, fungal and protozoan pathogens and discusses the convergently evolved commonalities as well as system-specific variations thereof. Key microbial molecules involved in host cell exit are highlighted and discussed as potential targets for future interventional approaches.
INTRODUCTION
Infectious diseases caused by viruses, bacteria, fungi, or parasites still represent a major cause of morbidity worldwide and account for almost 50,000 deaths every day [1]. During the course of infection, many bacterial, fungal and protozoan pathogens rely on a life-cycle phase, during which they parasitize host cells, typically but not always contained by membranous vacuoles. The intracellular life-style provides several advantages, including accessibility to nutrients and evasion from the human humoral and cellular immune system. Research during the last decades has revealed manifold examples illustrating the intricate and sophisticated strategies of microorganisms to invade their host cells and to manipulate the intracellular environment in order to promote microbial survival and to evade destruction by the host immune system (reviewed in [2]). Although crucial to ensure life-cycle progression, and, in consequence, dissemination and transmission, host cell exit has hitherto remained largely unresolved. Recent results, which were mainly gained from studies on Listeria, Toxoplasma and Plasmodium, provide firm evidence that host cell exit, initially viewed as a largely passive process, represents an active and dynamic interplay between the pathogen and the infected cell. Three major exit pathways, (i) the induction of programmed cell death (PCD), (ii) the active breaching of host cell-derived membranes, and (iii) the induced membrane-dependent exit without host cell lysis, have been identified. The pathways follow a spatially and temporally defined coordinated process and involve the interaction of pathogen- and host-derived effector molecules (reviewed in [3][4][5][6][7]).
–
Host cell exit by human pathogens is typically linked to tissue inflammation, organ dysfunction, and host-to-host transmission and thus significantly contributes to disease burden and the epidemiology of infectious diseases. Hence, a detailed knowledge of the underlying mechanisms of microbial cell exit is essential to understand the pathogenesis of infectious diseases. This is demonstrated by the critical role of exit-associated microbial effector molecules for the survival and spread of pathogens. An example is the recent finding that chemical inhibition of plasmepsin (PM) proteases can interrupt the egress of malaria parasites from the enveloping erythrocyte [8][9]. The present review therefore aims at attracting the well-deserved attention to this essential and understudied life-cycle phase of intracellular pathogens. We will explore the diverse exit strategies of intracellular-living bacterial, fungal, and protozoan pathogens, and discuss key microbial and host effector molecules. Further, we will assess the cellular and molecular aspects of microbial host cell exit and evaluate potential targets for future interventional strategies to combat infections by intracellular pathogens.
EXIT PATHWAYS
Current data indicate that at least three principal exit pathways are used by intracellular pathogens (outlined in Fig. 1) (reviewed in [3][4][5][6][7]): (i) PCD including the non-lytic apoptosis and the lytic necroptosis and pyroptosis pathways, employed by bacterial, fungal and protozoan pathogens; (ii) the active breaching of host cell-derived membranes such as the endosomal, the vacuolar and/or the host cell plasma membrane, as shown for a variety of bacterial and protozoan parasites; (iii) the induced membrane-dependent exit without host cell lysis, e.g. via actin-based protrusions, extrusions, budding, exocytosis, expulsion or ejection, as has been demonstrated for some bacteria, for the yeast Cryptococcus and for Plasmodium. While the first two pathways ultimately result in killing of the host cell, the third pathway in general leaves the host cell intact. Intriguingly, recent work suggests that the majority of intracellular pathogens is able to utilize more than one of these pathways, dependent on life-cycle stage, environmental factors and/or host cell type.
 | FIGURE 1: The three strategies of host cell exit. (1) Induction of programmed cell death, including the non-lytic apoptosis and the lytic necroptosis and pyroptosis pathways; (2) Active host cell destruction, comprising breaching of host cell membranes such as the vacuolar and/or the host cell plasma membrane; (3) Induced membrane-dependent exit without host cell destruction, e.g. by actin tail (yellow line)-mediated protrusions, extrusions, budding, exocytosis, expulsion or ejection via an ejectosome (yellow box). Pathogens for which distinct pathways were demonstrated are indicated. AB, apoptotic body; FC, fragmented chromatin; N, nucleus; P, pathogen; V, vacuole. |
–
Induction of programmed host cell death
PCD represents an intrinsic, regulated process of cell death with typical morphological changes and an important contribution to tissue ontogeny, cellular immunity and organ homeostasis. Different forms of PCD, i.e. apoptosis, necroptosis and pyroptosis, have been described that are initiated by specific signals and signal transduction cascades and that exhibit defined phenotypic characteristics (reviewed in [10]). PCD is also observed during infection, where it allows exfoliation and removal of infected cells. Here, a typical distinction is the one between lytic and non-lytic cell death, which has a profound impact of the immune response and, in consequence, on the course of infection. Lytic death is associated with early cell membrane permeabilization and release of cell debris, whereas non-lytic cell death maintains the surface integrity of the dying cell until its uptake by a phagocyte. Non-lytic cell death is largely confined to apoptosis whereas both pyroptosis and necroptosis cause early loss of plasma membrane integrity. Notably, even late stage apoptosis has a programmed lytic component, referred to as secondary necrosis. Accumulating evidence suggests that microbial targeting of PCD-inducing signal transduction pathways represents a cell exit strategy for a number of pathogens (Fig. 1).
–
Apoptosis
Apoptosis represents an evolutionarily conserved mechanism that contributes significantly to organ development and tissue homeostasis. Apoptosis also plays a prominent role in preventing pathogen progeny, as previously observed during viral infections [11]. It is characterized by typical morphological changes, including the disintegration of the apoptotic cell into condensed apoptotic bodies, which are subsequently taken up by phagocytes, particularly macrophages. Via uptake of these apoptotic bodies, viable pathogens could be transferred to a phagocytic cell in a non-inflammatory context. Apoptosis would therefore not release the intracellular bacterium to the extracellular space, but would rather lead to its re-uptake and thus cell-to-cell spread. The pathogen will remain within a membrane-enclosed particle and end up in a phagosome, where it has to deal with multiple degradative mechanisms. This is illustrated by the phagocyte-infecting gram-positive bacterium Mycobacterium tuberculosis, which is taken up within apoptotic bodies via efferocytosis [12]. M. tuberculosis is subsequently killed by the macrophage. In contrast, the protozoan parasite Leishmania spp., the causative agent of the vector-borne tropical disease leishmaniosis, is internalized by macrophages packed in apoptotic bodies and subsequently establishes itself in the phagosomal compartment [13][14]. Furthermore, the fungal pathogen Cryptococcus neoformans, which can cause infections of a number of organs, including the lung and central nervous system, can exit the cell through lytic and non-lytic mechanisms, and the lytic mechanism has been found to be of apoptotic nature, even though lysis does not normally occur during apoptosis [15].
–
Necroptosis
Necroptosis shows phenotypic characteristics of necrotic cell death (thus also termed programmed necrosis) and is induced by a specific signaling pathway that includes the receptor-interacting protein kinase 3 (RIP3/RIPK3) and the mixed lineage kinase domain like pseudokinase (MLKL) (reviewed in [16][17]). It has to be kept in mind that most mechanistic studies of necroptosis use agonists of death-receptor signalling to induce necroptosis. In this situation, necroptosis only occurs if caspase-8, which belongs to a group of PCD-related cysteine-aspartic proteases, is inhibited via chemical or gene-knock out approaches. Although such approaches have improved our understanding, this type of experimental design is not physiological. The role of necroptosis during infection therefore requires further investigations.
–
Necroptosis has been observed during infection with the facultative intracellular gram-negative bacterium Salmonella enterica serovar Typhimurium (S. Typhimurium), a pathogen causing gastroenteritis in humans following ingestion of contaminated food or water. Following penetration of the intestinal mucosal barrier, S. Typhimurium is taken up by macrophages and induces necroptosis [18]. Unexpectedly, this induction depends on type I interferon and the inhibition of necroptosis reduces the bacterial organ load [19]. However, it remains unresolved whether this is because cell exit is blocked or because macrophages survive and kill the bacterium.
–
A number of studies have investigated the role of necroptosis during mycobacterial infection. The most intriguing finding is that death of neutrophils that have engulfed virulent M. tuberculosis is instrumental to the transfer of the bacteria into macrophages, where they proliferate and induce persistent infection [20]. Cell death in neutrophils is induced via reactive oxygen species (ROS). Because the study was performed in human cells where the possibilities of genetic modification are limited, it is not clear though, if death of the neutrophil mechanistically represents necroptosis. As shown, chemical inhibition of neutrophilic ROS production prevents cell death. In addition, inhibition of RIPK1 (which is often involved in necroptosis) reduced bacterial transfer [20].
–
Another possible way of inducing necroptosis is through the cytosolic protein Z-DNA-binding protein ZBP1 (also known as DAI). Upon binding of specific conformations of nucleic acids, ZBP1 can activate the RIPK3/MLKL signalling axis [21]. While this has only been described for viruses so far [22], it would not be surprising if ZBP1 also had a function in bacterium- or parasite-induced PCD, given the overlap of viral and bacterial pattern recognition. In fact, ZBP1 was upregulated in M. tuberculosis-infected macrophages [23], although no specific function has been assigned.
–
Pyroptosis
Pyroptosis involves pores in the plasma membrane formed by members of the gasdermin family, following their proteolytic cleavage by caspases. Pyroptosis thus requires the proteolytic activity of at least one caspase and a number of caspases have been found to be able to induce gasdermin-cleavage and pyroptosis, e.g. caspase-1 and -11 (caspase-11 is only found in mice, its orthologues in humans are caspase-4 and -5), but also to some degree by the apoptotic caspase-3 [10][24][25][26]. These caspases are activated by multiprotein complexes known as inflammasomes that are formed by oligomerization of caspase-adapter proteins (reviewed in [27][28]).
–
Intriguingly, pyroptotic caspases can be activated upon recognition of microbial ‘patterns’ in the cytosol. An example is the recognition of cytosolic lipopolysaccharide by caspase-11 (reviewed in [28]). A substantial number of bacteria have been found to activate pyroptosis, among them Legionella, Francisella, Shigella, Salmonella and Listeria (reviewed in [3][4][7]). Previous investigations of pyroptosis during microbial infection have focused on its potential role in host defense rather than microbial host cell exit. The deletion of various components of pyroptosis signaling enhances the sensitivity against bacteria; the deletion of caspase-11 for instance sensitizes host cells and mice against enteric Salmonella infection [29] as well as against infection with Legionella pneumophila [30].
–
Another case in point is the opportunistic yeast pathogen Candida albicans, which induces pyroptosis in macrophages instrumental to facilitate release of the microbe [31]. It therefore seems very likely that pyroptosis, while involved in the antimicrobial host response and the initiation of inflammation, in addition plays a substantial role in cell exit. Pyroptosis is associated with secretion of proinflammatory cytokines that drive inflammation and pyroptotic PCD is expected to further augment inflammation through the release of both microbial components, making them available to the immune system and constituents of the dying cell (damage-associated molecular patterns; reviewed in [32]). Although inflammation is primarily a defense reaction with detrimental consequences to the pathogen, its downstream effects such as changes in the metabolism or influx of immune cells might actually favor growth and tissue spread of the pathogen. Microbe-directed skewing of the immune response by specific signals might further diminish the antimicrobial effect and enhance the pathogen’s benefit. Notably, pyroptosis, unlike apoptosis and probably necroptosis, is limited to specific cell types, such as macrophages, since not all cells express or can activate inflammasome components.
–
Active host cell destruction
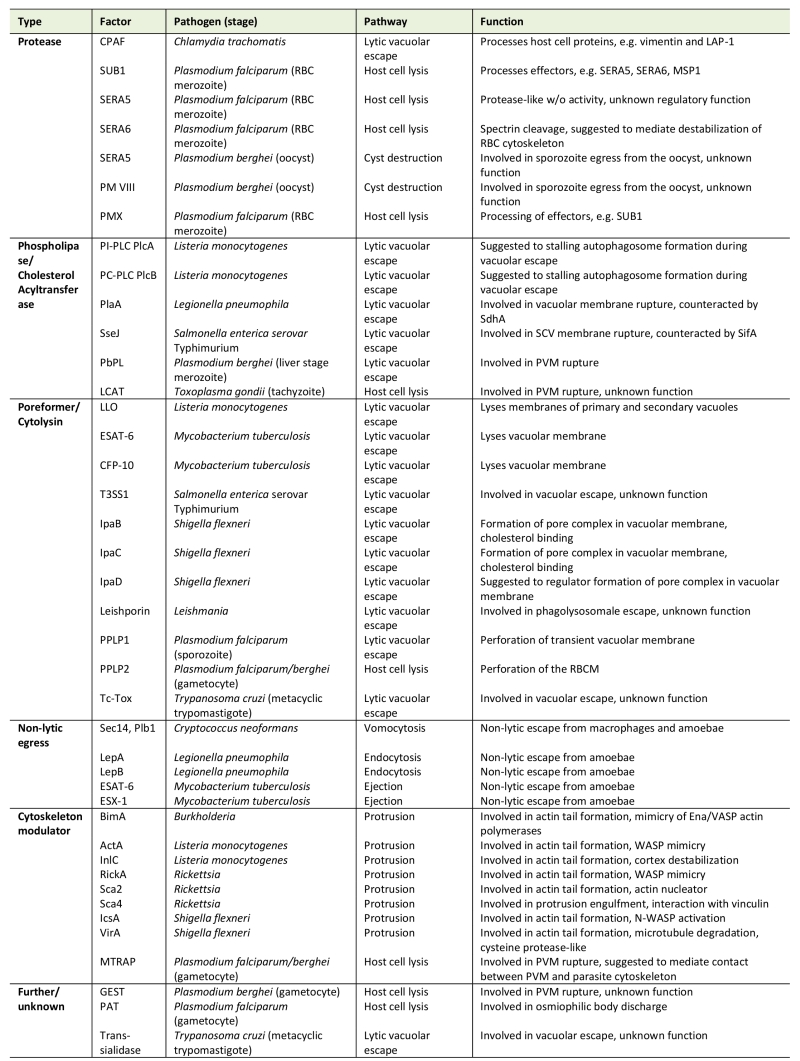
Active host cell destruction describes an exit process, during which microbial molecules penetrate or perforate membranes, such as the host cell plasma membrane or the membrane of the vacuolar compartment, in consequence destroying host cell and compartment, respectively (Fig. 1). Exceptionally, active destruction can include the walls of intra- or extracellular pathogen-containing cysts. Particularly, three types of proteins were described that mediate active host cell lysis, i.e. proteases, phospholipases, and pore-forming proteins (Table 1).
–
Host cell lysis
Active host cell lysis, which includes the destruction of both host cell plasma membrane and vacuolar membrane, is typical for Apicomplexan parasites and has best been studied for the intraerythrocytic blood and gametocyte stages of malaria parasites, particularly of P. falciparum, the causative agent of malaria tropica (Fig. 2). These stages reside within a parasitophorous vacuole (PV) inside red blood cells (RBCs). Following replication in the RBCs, the generated merozoites ultimately have to escape these to disseminate. RBC exit by merozoites follows an inside-out mode, meaning the PV membrane (PVM) ruptures prior to the RBC membrane (RBCM) (reviewed in [6]). The egress cascade begins with the activation of the plasmodial cGMP-dependent protein kinase G (PKG) by a yet unknown signal [33]. Concurrently, the calcium-dependent protein kinase CDPK5 becomes activated due to an increase of intracellular calcium [34][35]. The concerted activation of both kinases triggers a protease cascade that mediates merozoite egress. Specialized secretory organelles, called exonemes, discharge their content into the PV lumen, including the subtilisin-like protease SUB1 [36]. SUB1 has several targets, like the multiprotein merozoite surface complex called MSP1/6/7 and members of the soluble papain-like PV-resident SERA (sera-repeat antigen) proteins. The aspartic protease PMX, which is also located in the exonemes, mediates the proteolytic maturation of SUB1 [8][36][37][38][39]. In the PV, SERA5 and SERA6 as well as the merozoite surface-resident 200-kDa protein MSP1 become processed by SUB1 [40][41][42]. Within minutes following the increase of intracellular calcium, the PVM ruptures by a yet unknown mechanism and the RBCM is perforated [43][44][45][46]. The processed SERA6, which has β-spectrin cleaving activity, promotes destabilization of the RBC cytoskeleton, while SERA5 exhibits additional regulatory functions [41][42][46]. Eventually, the RBCM ruptures at a single breaking point and curls back, a rapid process that disperses the merozoites [47][48]. Interestingly, merozoite egress from the RBC does not involve actin-myosin motor-driven motility, contrary to sporozoite egress from the oocyst (see below) [49]. RBC exit by merozoites can be inhibited by cysteine and serine protease inhibitors. These include the recently identified PMX-targeting aminohydantoins as well as the hydroxyl-ethyl-amine-based compound 49c, which targets PMIX in addition to PMX [8][9].
 | FIGURE 2: Active host cell lysis by Apicomplexan parasites. The key molecules and the sequence of action during host cell egress by merozoites and gametocytes of Plasmodium and by tachyzoites of Toxoplasma gondii, respectively, are indicated. Question marks indicate steps with ambiguous or unknown interactions. DG, dense granule; EV, egress vesicle; EX, exoneme; HCM, host cell membrane; MN, microneme; N, nucleus; OB, osmiophilic body; PVM, parasitophorous vacuole membrane; RBCM, red blood cell membrane. |
–
Egress of gametocytes from the RBC occurs at the onset of gametogenesis, once the plasmodia have been taken up by a blood-feeding mosquito, and also follows an inside-out mode. In the mosquito midgut, the gametocytes are activated by external stimuli, particularly a drop in temperature and the contact with the mosquito-derived molecule xanthurenic acid (XA) (reviewed in [50][51][52]). While a plasmodial receptor that binds XA has not been identified, it was shown that gametocyte activation leads to cGMP synthesis and PKG activation [53][54]. PKG controls the synthesis of phosphatidylinositol-(4,5)-bisphosphate (PIP2) via phosphorylation of the phosphoinositol kinases PI4K and PIP5K [55]. A phosphoinositide-specific phospholipase PI-PLC is further activated during the egress cascade, leading to the hydrolysis of PIP2 into diacylglycerol (DAG) and inositol-(1,4,5)-trisphosphate (IP3) [56]. IP3 is suggested to be responsible for the opening of calcium channels in the endoplasmic reticulum, although no plasmodial orthologue of an IP3 channel has been identified so far. The calcium ions then regulate two kinases, CDPK1 and CDPK4, which induces the initiation of DNA synthesis in the replicating male gametes and the release of translational repression in female gametes [57][58]; these events mark the onset of male and female gametogenesis, respectively.
–
At least two types of vesicles are discharged during gametocyte egress from the RBC. During the first minutes following gametocyte activation, the osmiophilic bodies release their content into the PV lumen. These contain a variety of egress-related proteins, like Pg377, MDV-1/Peg3, the subtilisin protease SUB2, the dipeptidyl aminopeptidase DPAP2, PMVI, the putative pantothenate transporter PAT and the gamete egress and sporozoite traversal protein GEST [59][60][61][62][63][64][65][66][67]. Consequently, the PVM ruptures at multiple sites. Although the precise mechanism of PVM rupture is undefined, it appears to involve the merozoite thrombospondin-related adhesive protein MTRAP [64][65][68]. It was suggested that the membrane-spanning MTRAP links the gametocyte plasma membrane and the PVM and needs to be dismantled prior to PVM rupture, and indeed a rhomboid-protease cleavage site was identified in MTRAP [69]. In a subsequent calcium-dependent step, a second set of vesicles is released into the RBC cytoplasm. These ‘egress vesicles’, which might represent a sub-type of osmiophilic bodies, contain the plasmodial perforin-like protein PPLP2 [63][64]. Upon discharge, PPLP2 perforates the RBCM, resulting in the release of the RBC cytoplasm [70][71]. Noteworthy, the male and female gametes that have meanwhile formed are still contained in the remnants of the RBCM for several more minutes, before the RBCM opens via a single pore to release the fertile gametes [71][72][73]. The delayed cell exit might represent a way to circumvent the host’s immune system. Because RBCM breakdown is sensitive to inhibitors of cysteine and serine proteases, it was suggested that the enzymatic cleavage of RBC cytoskeletal proteins precedes membrane rupture [72][74].
–
Host cell exit has further been investigated for the Apicomplexan parasite Toxoplasma gondii, a feline pathogen that, if taken up by humans, can infect various tissues and causes severe toxoplasmosis, particularly in immunocompromised individuals (Fig. 2). The lecithin-cholesterol acyltransferase LCAT of T. gondii was reported to be released from dense granules, where it associates with the parasite plasma membrane as well as with the PVM prior to egress, suggesting a role in either mediating microneme release or PVM break-down [75][76]. In addition, the microneme-resident perforin-like protein PLP1 is secreted into the PV, which mediates PVM rupture by forming hexameric pore complexes [77][78][79]. The micronemes play a central role in host cell exit by T. gondii and hence molecules involved in microneme maturation and functionality, e.g. the phosphoglucomutase-related proteins, the micronemal protein MIC2, or the secretory protein ASP3 [80][81][82], affect parasite egress. Micronemal discharge is stimulated by a variety of factors, which include acidification, serum albumin and the reduction of potassium levels in the host cell cytoplasm [83][84][85]. Downstream, a PKG becomes activated, in turn regulating PI-PLC activity, which results in increased cytosolic calcium levels [86]. PKG activity is regulated amongst others via cross talk with the protein kinase PKA. Calcium levels appear to be further regulated by the second messenger cyclic ADP ribose (cADPR) via a yet unknown endoplasmic reticulum receptor [87][88]. It was suggested that cADPR synthesis itself is controlled by the endogenous stress regulator abscisic acid (ABA) [89]; however, neither an ABA receptor nor the signaling pathway induced by ABA has hitherto been identified. In addition, calcium homeostasis is controlled by phosphatidylthreonine (PtdThr) levels, and abrogation of PtdThr synthesis results in the motility-dependent egress of T. gondii [90][91][92]. The increased calcium levels activate the kinases CDPK1 and CDPK3. While CDPK1 is required for microneme secretion, CDPK3 facilitates rapid initiation of motility during parasite egress by phosphorylation of myosin and the suppressor of calcium-dependent egress 1 protein [84][93][94][95][96]. Also involved in micronemal secretion is DAG and its downstream product phosphatidic acid, which is recognized by the microneme-associated acetylated pleckstrin-homology domain-containing protein APH prior to microneme secretion [97].
Table 1. Microbial key factors of host cell exit. |
 |
–
Lytic vacuolar escape
Exit via active destruction has also been described for the escape from the vacuolar compartment by a variety of pathogens, even if the final exit mode follows another route. For example, the plasmodial liver stages use a secreted phospholipase (PbPL) to disrupt the PVM (although the release from infected hepatocytes eventually occurs by budding; see section Extrusion and budding) [98]. Noteworthy in this context, prior to settling down in a host hepatocyte contained in a PV, the sporozoites traverse through a variety of tissue cells. Cell traversal initially occurs within a transient (non-parasitophorous) vacuole, which the sporozoite subsequently escapes, a process requiring the the plasmodial perforin PPLP1 [99][100].
–
Furthermore, the intracellular Kinetoplastida parasites Leishmania spp. and Trypanosoma cruzi, which mainly parasitize human phagocytes, are initially contained in a vacuolar compartment of phagosomal and lysosomal origin, respectively, before they escape into the host cell cytoplasm. Their vacuolar escape mechanisms involve parasite-derived molecules. For example, L. amazonensis expresses leishporin, a protein with membrane-lytic activity [101][102]. However, a direct proof of active vacuolar lysis by leishporin has not been provided [103]. In addition, the bloodstream trypomastigotes of T. cruzi express a trans-sialidase on their surface, which might mediate vacuolar escape my removing sialic acid from the vacuolar membrane [104][105]. Further, trypomastigotes secrete a pore-forming hemolytic toxin Tc–TOX into the vacuole, which is activated by the acidic pH of the lysosomal compartment. It has been proposed that the concerted action of the two proteins mediates rupture of the lysosomal membrane [106][107]. The detailed mode of action of Tc-TOX, though, is still unknown.
–
The vacuolar escape of some intracellular bacteria occurs immediately following uptake to evade endosome-lysosome fusion and allow replication within the host cell cytosol. This has been shown e.g. for Shigella, Listeria, Francisella, and Rickettsia. In contrast, bacteria like Legionella, Mycobacterium, and Chlamydia that remain within the endosomal vacuole manipulate this compartment to inhibit phagosomal-lysosomal fusion. These bacteria have adapted to this modified endosomal compartment and escape the vacuole only shortly before host cell exit (reviewed in [108]). For some of these bacterial pathogens, cytolytic proteins and phospholipases involved in active vacuolar membrane lysis have been reported.
–
Lytic vacuolar escape has been investigated in detail for the facultative intracellular gram-positive bacterium Listeria monocytogenes, which is able to infect a variety of cells including phagocytes. While L. monocytogenes enters the host cell within a primary vacuole, the bacterium rapidly escapes the vacuole in order to replicate in the cytosol. Vacuolar escape is essential for intracellular growth, because L. monocytogenes replicates inefficiently when contained in vacuoles [109][110][111]. Cytosolic L. monocytogenes uses actin-based motility to spread to the neighboring cells (see section Actin-mediated protrusion). Following cell-to-cell transit, L. monocytogenes resides in a vacuole enclosed by two membranes, one layer derived from the donor and one from the recipient cell. L. monocytogenes also rapidly escapes this secondary vacuole and continuous replicating in the cytosol [112]. To allow endosomal escape, L. monocytogenes expresses a pore-forming cholesterol-dependent cytolysin, termed listeriolysin O (LLO), which mediates the escape from both, the primary and the secondary vacuoles [113]. Additionally, two phospholipases, PlcA and PlcB, contribute to endosomal membrane disruption [[114][115]; reviewed in [116]). The two enzymes additionally function in subverting host autophagic defenses by stalling autophagosome formation [117][118]. The LLO pore-forming activity is enhanced by phagosomal acidification [119][120][121].
–
Potential membrane-lytic functions have further been assigned to the type III-secreted translocator proteins IpaB and IpaC of the gram-negative rod Shigella flexneri, which causes shigellosis in humans [122][123][124]. The two proteins form a complex in the vacuolar membrane that binds cholesterol, resulting in membrane degradation. A third protein, IpaD, seems to have regulatory function during this process [125]. Vacuolar escape has also been reported for mycobacteria [126][127]. Two secreted membrane-lytic proteins, ESAT-6 (a 6-kDa early secretory antigenic protein) and CFP-10 (a 10-kDa culture filtrate protein), were assigned to this process [128][129][130][131][132]. Both proteins are exported into the host cell cytosol through the type VII secretion system ESX-1 (reviewed in [133][134]); their exact mode of action, however, is still unclear. Vacuolar escape of mycobacteria rapidly leads to the initiation of PCD (see section Necroptosis) [135][136].
–
Chlamydia trachomatis is an obligate intracellular gram-negative bacterium that infects epithelial cells of the urogenital tract and the conjunctivae. In host cells, the bacteria are contained in vacuolar compartments termed inclusions. C. trachomatis can exit the host cell by either extrusion (see section Extrusion and budding) or by lysis. Here, C. trachomatis first lyses the inclusions by a yet unknown mechanism to escape into the host cell cytoplasm. In preparation for escape, C. trachomatis translocates virulence factors into the host cell cytoplasm, among others via a type III secretion system (T3SS). The chlamydial protease-like activity factor CPAF, a serine protease, is initially secreted into the inclusion lumen and eventually crosses the inclusion membrane [137]. Following its release into the cytosol, CPAF processes a wealth of host proteins to promote chlamydial intracellular growth, particularly vimentin and the lamin-associated protein LAP-1 [138]. Chlamydiae lacking CPAF are unable to egress from the host cell [132]. Host cell exit by C. trachomatis further requires a variety of plasmid-encoded virulence factors like the transcription regulator Pgp4 [139]. Since cell lysis can be blocked by the cysteine protease inhibitor E64, it has been suggested that cytoskeleton degradation precedes rupture of the host cell membrane [140].
–
In epithelial cells of the intestine, S. Typhimurium resides in a membrane-bound endosome, termed Salmonella-containing vacuole (SCV), where it is able to proliferate [141][142][143]. During the first hours post-invasion, bacteria of a minor proportion of infected cells escape into the cytosol, where they rapidly replicate. The SCV biogenesis involves numerous bacterial effector molecules translocated by the type III secretion systems 1 and 2 (T3SS1 and T3SS2, respectively) (reviewed in [144][145]). While cytosolic S. Typhimurium exhibit T3SS1 expression, T3SS2-positive bacteria remain in the SCV [146]. Noteworthy, pore-forming activities were demonstrated for T3SS1 mediated effector molecules and it is thought that the T3SS1 plays a role in damaging the nascent (early) SCV [147][148][149][150]. In addition, several T3SS2-translocated effectors like the protein Salmonella-induced filaments SifA and SseJ homologous to L. pneumophila PlaA (see below) are involved in SCV stabilization and destabilization, respectively, at later time points. SifA counteracts the phagosome rupture by SseJ, which shows PLA and cholesterol acyltransferase activities [151][152][153]. SifA binds to SKIP (SifA and kinesin-interacting protein), which in turn interacts with kinesin-1 and the secreted effector protein PipB2. This interaction might regulate SCV stability [154][155][156].
–
Legionella pneumophila, a facultative intracellular bacterium, infects amoeba under environmental conditions. Once entering the human lung, it can invade macrophages, thereby causing severe lung infections. In both, macrophages and amoebae, L. pneumophila replicates in a specialized phagosome avoiding fusion with the host endocytic pathway. A type IVB secretion system (Dot/Icm) responsible for the translocation of a multitude of effector proteins into the host cell as well as the type II secretion system Lsp are required for intracellular replication (reviewed in [157]). L. pneumophila exits from the phagosome either by non-lytic exocytosis (see section Exocytosis, expulsion and ejection) or via a pore-forming activity [158][159][160][161]. Pore formation might be triggered by phospholipases found in L. pneumophila (reviewed in [162]). Specifically, the type II secreted lysophospholipase PlaA, showing homology to S. Typhimurium SseJ (see above), destabilizes the phagosomal membrane in the absence of the type IVB-secreted protective factor SdhA [163][164]. The detailed mode of phagosome lysis remains to be investigated.
–
A special form of lysis-mediated microbial exit is the one from cysts. This has been investigated for the egress of P. berghei from oocysts, parasite-derived cyst-like structures, which form after sexual reproduction and are found in the mosquito midgut attached to the epithelium. Oocyst exit required two proteases, P. berghei SERA5 (an orthologue of P. falciparum SERA8) and PMVIII [165][166]. Other plasmodial proteins that mediate sporozoite maturation, e.g. the thrombospondin-related protein TRP1 or the LCCL-domain containing proteins, were further assigned to oocyst egress by malaria parasites [167][168][169][170]. Noteworthy, the motility of Plasmodium sporozoites, driven by an actin-myosin motor, is necessary for oocyst egress [170].
–
Induced membrane-dependent exit
Some microbes are able to leave the intact host cell. Such exit strategies include actin-driven membrane protrusions enabling the spread of single bacteria between cells, extrusions and budding of microbes packed in a membranous compartment as well as ejection, expulsion and exocytosis of free microbes (Fig. 1). The detailed mechanisms of induced membrane-dependent exit, however, are to date not well defined and only few key molecules of this exit pathway have been identified (Table 1).
–
Actin-mediated protrusion
Polar recruitment and polymerization of actin results in a directed locomotion of cytosolic bacteria. Actin polymerization is facilitated by the expression of a microbial surface protein that binds or mimics the host cell actin-related protein (Arp)2/3 complex [171]. Reaching the cell border, the advancing bacterium is able to protrude the host cell plasma membrane by physical force. Since bacteria are unable to penetrate this membrane, they project from the infected cell within the tip of a filopodium-like membrane extension. This cell membrane protrusion is subsequently engulfed by the adjacent cell [172]. Notably, this cell-to-cell spread occurs without contact to the extracellular environment and thus protects the pathogen from exposure to extracellular immune surveillance mechanisms and antimicrobial effector molecules. The most intensively studied microorganisms performing actin-mediated protrusion are Listeria monocytogenes and Shigella flexneri, but a similar ability has been demonstrated for the spotted fever group of rickettsiae, Burkholderia spp., Ehrlichia spp. and Mycobacterium marinum.
–
Upon release from the endosomal membrane, L. monocytogenes starts polar expression of the surface molecule actin assembly-inducing protein (ActA) [173]. The N-terminus of ActA represents a mimic of the naturally occurring actin nucleation-promoting factor Wiscott-Aldrich syndrome protein (WASP) and binds preformed Arp2/3 complexes [174]. The central domain of ActA additionally binds profilin and vasodilator-stimulated phosphoprotein (VASP), promoting the rapid formation of branched actin polymers. Furthermore, parallel actin polymerization and actin tail assembly is facilitated by the activation of Rho GTPase and the actin polymerization factor formin [112][175]. The actin comet tail thereby propels the bacterium through the host cell cytosol. Once approaching the vicinity of the plasma membrane the secreted Listeria protein internalin C (InlC) binds to the host adaptor protein Tuba to inhibit the actin polymerization promoting neural WASP (N-WASP) and to the COPII complex component SEC31 [176]. This weakens the cortical tension at the plasma membrane allowing Listeria to protrude the plasma membrane in a filopodia-like fashion [175]. The membrane cytoskeleton linker ezrin accumulates at the protrusion site and stabilizes the growing membrane extension [177]. While it is still unclear, how exactly the bacterium-containing membrane extension is engulfed by the adjacent cell, the process requires active participation of both, the donor and recipient cell. For example, the host casein kinase CSNK1A1 was shown to promote the conversion of protrusions to endosomes in the donor cell [178][179]. After transfer, Listeria resides in an endosomal compartment composed of a membrane layer of both the donor and recipient cell. This double-layered endosomal membrane is lysed by PlcB, PlcA, and LLO releasing the bacterium in the recipient cell cytosol (see section Lytic vacuolar escape) [115][180][181][182][183]. Importantly, the ability to perform cell-to-cell spread represents a critical component of L. monocytogenes virulence and does not lead to host cell death [181][184][185].
–
Actin-based motility also critically contributes to Shigella flexneri virulence [186]. Here, the Shigella IcsA protein activates N-WASP to recruit Arp2/3 and initiate actin tail formation [187][188][189]. Simultaneously, Shigella stimulates RhoA GTPases and the mammalian diaphanous-related formins mDia1 and mDia2 to promote actin polymerization in parallel arrays at the protrusion site [190]. The virulence factor VirA of S. flexneri degrades the cytoplasmic microtubule network via its cysteine protease-like activity and might thereby promote cytosolic locomotion [191][192]. Similar to Listeria, engulfment of the Shigella-containing protrusions by the adjacent cell does not depend on additional bacterial factors. Instead, it requires active participation of the host cell proteins. The requirement for tricellulin, epsin-1, clathrin and dynamin-2 suggest the involvement of a noncanonical clathrin-dependent endocytic pathway in the recipient cell [193][194].
–
Other bacteria that employ actin-based motility to allow cell-to-cell transfer include the spotted fever group rickettsiae such as Rickettsia rickettsii, R. conorii or R. parkeri [195]. The molecular basis, however, is less well examined. Early motility has been attributed to polar expression of RickA, which recruits the Arp2/3 complex and induces actin polymerization in an N-WASP-like manner. Later, motility occurs in an Arp2/3 independent fashion via secretion of the autotransporter surface cell antigen (Sca) 2, a formin-like actin nucleator that generates long unbranched actin tails [196][197]. Subsequent secretion of Sca4 promotes protrusion engulfment through interaction with the cell-adhesion protein vinculin. This relieves intercellular tension and the engulfment of bacteria-containing protrusions [198].
–
Ehrlichia chaffeensis recruits N-WASP to generate actin polymerization and form bacteria-driven filopodia and allow cell-to-cell spread [199][200]. The pathogenesis of Burkholderia spp. relies on actin-based motility [201]. Whereas the secreted trimeric autotransporter BimA (Burkholderia intracellular motility A) of the animal pathogen B. thailandensis activates the host Arp2/3 complex, the orthologue BimA of the human-pathogenic B. pseudomallei and B. mallei mimic host Ena/VASP actin polymerases [202]. In contrast to the above-discussed pathogens, cell-to-cell spread is facilitated by fusion of infected and adjacent cells induced by the type VI secretion system (T6SS)-1 [203]. Mycobacterium marinum, a natural fish pathogen and occasional human pathogen, requires either WASP or N-WASP to perform actin-based motility and both the Arp2/3 complex and the vasodilator-stimulated phosphoprotein were identified as constituents of the actin tail [126][204]. The molecular mechanism of subsequent cell-to-cell spread may differ from the other pathogens.
–
Extrusion and budding
Host cell exit via extrusion or budding involves the release of membrane-encircled microorganisms (Fig. 1). Here, the membrane coat protects the microbe against humoral factors of the host immune system. To date, only few molecular players have been identified that modulate this type of host cell exit (Table 1).
–
Extrusions have been reported for Chlamydia trachomatis. Chlamydial extrusions depend on actin polymerization mediated by N-WASP and Rho GTPases, while myosin and septins are involved in regulation and stabilization of the actin filaments [140][205]. A bacterial protein appears to recruit the myosin-activating machinery to the inclusion to favor extrusion of pathogens over a cell-lytic pathway [206].
–
Viral-like budding was shown for the scrub typhus-causing bacterium Orientia tsutsugamushi that replicates in the cytosol of a variety of host cells, including phagocytes. Inside its host cell, the bacterium is ‘encapsulated’ by the plasma membrane of the cell. Host cell exit appears to depend on lipid rafts and a bacterial protein that was found to co-localize with caveolins at the site of cell exit, suggesting a role in egress ([207], reviewed in [208]).
–
Host cell exit by budding was further demonstrated for the intrahepatic merozoites of Plasmodium. Following phospholipase-mediated PV breakdown (see section Lytic vacuolar escape), these lie in the hepatocyte cytoplasm and are subsequently released into the blood stream. The merozoites leave the hepatocyte in host cell plasma membrane-derived vesicles termed merosomes that can contain a few up to several hundreds of merozoites [209]. The parasites induce the separation of the actin cortex from the hepatocyte plasma membrane prior to merozoite formation [210]. The molecular mechanisms that allow the vesicle to bud from the hepatocyte and penetrate the endothelial cell layer to reach the blood stream are, however, unknown. Interestingly, although PCD is induced by the parasite in the infected hepatocyte, the budding merosomes do not expose phosphatidylserine on their surface as would be expected from apoptotic cells [211]. This probably helps the merosomes to remain undetected by macrophages as they leave the liver and travel to the lung tissue [212]. Contrary to the induced membrane-dependent exit by other intracellular pathogens, in this case, the host hepatocyte dies.
–
Exocytosis, expulsion and ejection
Exocytosis involves the transport of molecules from intracellular endosomal vesicles to the plasma membrane. The membrane of transport vesicles fuses with the plasma membrane to allow cargo release and thereby aids the passage of mostly large and polar substances into or through the plasma membrane (reviewed in [213][214]). A variety of pathogenic microbes exit host cells by exocytosis and exocytosis-like expulsion (also called vomocytosis) as well as by mechanistically distinct ejection. All these procedures have in common that they release free pathogens without harming the host cell. The processes therefore avoid the release of proinflammatory cellular constituents (reviewed in [4][5][215]).
–
Exocytosis-like egress from amoebae has been described for Legionella pneumophila. Within the host cell, L. pneumophila is contained in a specialized phagosome and via a type IVB secretion system secretes effector proteins into the host cell cytoplasm to allow replication (see section Lytic vacuolar escape) (reviewed in [157]). Such effectors frequently harbor signatures of eukaryotic proteins and some of these exert homology to the SNARE (soluble N-ethylmaleimide-sensitive factor attachment protein receptor) proteins known to mediate the fusion of vesicles to biological membranes [216][217][218][219]. These analogous bacterial effectors may drive or even abrogate vesicle fusion events. Two type IVB-secreted L. pneumophila proteins, LepA and LepB, with regions weakly similar to mammalian early endosome antigen 1 (EEA1), required for endosome docking of SNARE proteins, were identified. They play a critical role in the bacterial release from the amoebae Acanthamoeba castellanii or Dictyostelium discoideum [216], indicating that L. pneumophila is released from amoebae by an exocytotic pathway. However, earlier reports described vesicle-associated Legionella expelled from A. castellanii [220]. This raises the question whether the bacteria are indeed released by the above-described process in their free form or rather in a vesicle-enclosed form.
–
Non-lytic egress of the fungus Cryptococcus neoformans is debated as an important factor determining systemic dissemination of the pathogen [221][222]. The yeast exerts an exocytosis-like expulsion, called vomocytosis, in macrophages. Non-lytic expulsion of Cryptococcus occurs through fusion of the phagosome and plasma membrane. Mutants defective for the secreted phospholipase PLB1 or the PLB1-exporting secretion system Sec14 exhibit reduced quantities of vomocytosis. The actin cytoskeleton of the host cell is not essential for this process [223][224][225][226]. However, macrophage phagosomes containing intracellular cryptococci undergo repeated cycles of actin polymerization, called actin ‘flashes’ dependent on the classical WASP-Arp2/3 complex. Prior to expulsion, the majority of phagosomes is permeabilized, which is immediately followed by an actin flash likely devoted to temporarily inhibit expulsion [225]. Vomocytosis has also been observed upon in vivo infections of mice and zebrafish [227][228]. The latter study reported that inhibition of the mitogen-activated protein kinase ERK5 increased vomocytosis and decreased pathogen dissemination, indicating that vomocytosis enhancement might represent a therapeutic target.
–
A particular mode of cell exit was discovered in Mycobacterium tuberculosis and M. marinum. Both are non-lytically ejected from D. discoideum through an F-actin-based structure, the ejectosome [229]. The process involves the so-called region of difference RD1 locus, where components of the mycobacterial type VII secretion system ESX-1 are encoded [127][130][135][229][230]. Both the ESX-1 secretion system and its secreted effector ESAT-6/EsxA are required for mycobacterial ejection from Dictyostelium and bacterial translocation into the cytosol of mammalian cells. A recent report describes an unexpected role of the autophagic machinery in non-lytic release of Mycobacteria and cell-to-cell transmission in Dictyostelium [231]. Further, the extraordinary importance of maintaining membrane integrity during the process of ejection is highlighted. Specifically, the study indicates that bacteria shortly prior to ejection are escorted by an autophagocytic vacuole, which is recruited in an ESX-1 independent manner. When autophagy is impaired, cell-to-cell transmission is inhibited. In this case, the host plasma membrane becomes compromised and the host cell subsequently dies [231]. These findings illustrate that non-lytic egress by ejection requires host cell-derived membrane protection pathways.
HOST CELL EXIT BY INTRACELLULAR PATHOGENS: THE ACHILLES’ HEEL?
The combined data highlighted in this review suggest that host cell exit by intracellular pathogens represents a fundamental and active step in infection, which, shaped by evolutionary pressure, is crucial for microbial spread and might represent the Achilles´ heel of microbial pathogenesis. A limited set of host cell exit pathways appears to be shared by a high variety of phylogenetically different microbes with the involvement of similar types of pathogen-derived proteins, like proteases, pore-forming proteins and phospholipases or actin-binders. This strongly suggests convergent evolution of the exit machineries. The fact that exit strategies employ manipulated secretion or delivery routes as well as the complex cytoskeletal restructuring further point to the intimate interaction between pathogens and their host cells.
–
While much experimental work lies ahead of us to decipher the molecular mechanisms of host cell exit by intracellular pathogens, evidence emerges that key molecules of host cell exit are promising targets for novel types of interventional strategies. The fact that only few classes of pathogen-derived proteins appear to be involved in the exit processes and that in general their accessibility for inhibitors is known makes host cell exit as a point of attack even more attractive. Indeed, as described earlier, first protease inhibitors have been identified that are able to block the egress of malaria parasites from RBCs [8][9]. While targeting microbial exit might not protect from primary infection, the cell-entrapment of microbes ensures immediate control of microbial tissue spread and disease progression. Importantly, it should still allow stimulation of a protective adaptive immune response thus providing therapeutic and prophylactic advantages. Such approaches have been exploited in the recent past for liver stage-targeting antimalarial vaccines, using attenuated parasites (reviewed in e.g. [232][233][234]) and might represent a pioneering strategy to combat life-threatening human infectious diseases. Concluding, present pieces of evidence point to exit strategies of intracellular pathogens as an emerging field of infection biology essential to fully understand and successfully counteract microbial pathogenesis.
References
- . Wolrd Health Organization, "World health statistics 2018", Available: http://apps.who.int/iris/bitstream/handle/10665/272596/9789241565585-eng.pdf. Accessed 26.06.2018, 2018.
- U. Schaible, and A. Haas, " Intracellular niches of microbes - A pathogens´ guide through the host cell.", Wiley-VCH, Hoboken., 2009.
- K. Hybiske, and R.S. Stephens, "Exit strategies of intracellular pathogens", Nature Reviews Microbiology, vol. 6, pp. 99-110, 2008. http://dx.doi.org/10.1038/nrmicro1821
- K. Hybiske, and R. Stephens, "Cellular Exit Strategies of Intracellular Bacteria", Microbiology Spectrum, vol. 3, 2015. http://dx.doi.org/10.1128/microbiolspec.VMBF-0002-2014
- N. Friedrich, M. Hagedorn, D. Soldati-Favre, and T. Soldati, "Prison Break: Pathogens' Strategies To Egress from Host Cells", Microbiology and Molecular Biology Reviews, vol. 76, pp. 707-720, 2012. http://dx.doi.org/10.1128/MMBR.00024-12
- C.C. Wirth, and G. Pradel, "Molecular mechanisms of host cell egress by malaria parasites", International Journal of Medical Microbiology, vol. 302, pp. 172-178, 2012. http://dx.doi.org/10.1016/j.ijmm.2012.07.003
- A. Traven, and T. Naderer, "Microbial Egress: A Hitchhiker's Guide to Freedom", PLoS Pathogens, vol. 10, pp. e1004201, 2014. http://dx.doi.org/10.1371/journal.ppat.1004201
- A.S. Nasamu, S. Glushakova, I. Russo, B. Vaupel, A. Oksman, A.S. Kim, D.H. Fremont, N. Tolia, J.R. Beck, M.J. Meyers, J.C. Niles, J. Zimmerberg, and D.E. Goldberg, "Plasmepsins IX and X are essential and druggable mediators of malaria parasite egress and invasion", Science, vol. 358, pp. 518-522, 2017. http://dx.doi.org/10.1126/science.aan1478
- P. Pino, R. Caldelari, B. Mukherjee, J. Vahokoski, N. Klages, B. Maco, C.R. Collins, M.J. Blackman, I. Kursula, V. Heussler, M. Brochet, and D. Soldati-Favre, "A multistage antimalarial targets the plasmepsins IX and X essential for invasion and egress", Science, vol. 358, pp. 522-528, 2017. http://dx.doi.org/10.1126/science.aaf8675
- L. Galluzzi, I. Vitale, S.A. Aaronson, J.M. Abrams, D. Adam, P. Agostinis, E.S. Alnemri, L. Altucci, I. Amelio, D.W. Andrews, M. Annicchiarico-Petruzzelli, A.V. Antonov, E. Arama, E.H. Baehrecke, N.A. Barlev, N.G. Bazan, F. Bernassola, M.J.M. Bertrand, K. Bianchi, M.V. Blagosklonny, K. Blomgren, C. Borner, P. Boya, C. Brenner, M. Campanella, E. Candi, D. Carmona-Gutierrez, F. Cecconi, F.K. Chan, N.S. Chandel, E.H. Cheng, J.E. Chipuk, J.A. Cidlowski, A. Ciechanover, G.M. Cohen, M. Conrad, J.R. Cubillos-Ruiz, P.E. Czabotar, V. D’Angiolella, T.M. Dawson, V.L. Dawson, V. De Laurenzi, R. De Maria, K. Debatin, R.J. DeBerardinis, M. Deshmukh, N. Di Daniele, F. Di Virgilio, V.M. Dixit, S.J. Dixon, C.S. Duckett, B.D. Dynlacht, W.S. El-Deiry, J.W. Elrod, G.M. Fimia, S. Fulda, A.J. García-Sáez, A.D. Garg, C. Garrido, E. Gavathiotis, P. Golstein, E. Gottlieb, D.R. Green, L.A. Greene, H. Gronemeyer, A. Gross, G. Hajnoczky, J.M. Hardwick, I.S. Harris, M.O. Hengartner, C. Hetz, H. Ichijo, M. Jäättelä, B. Joseph, P.J. Jost, P.P. Juin, W.J. Kaiser, M. Karin, T. Kaufmann, O. Kepp, A. Kimchi, R.N. Kitsis, D.J. Klionsky, R.A. Knight, S. Kumar, S.W. Lee, J.J. Lemasters, B. Levine, A. Linkermann, S.A. Lipton, R.A. Lockshin, C. López-Otín, S.W. Lowe, T. Luedde, E. Lugli, M. MacFarlane, F. Madeo, M. Malewicz, W. Malorni, G. Manic, J. Marine, S.J. Martin, J. Martinou, J.P. Medema, P. Mehlen, P. Meier, S. Melino, E.A. Miao, J.D. Molkentin, U.M. Moll, C. Muñoz-Pinedo, S. Nagata, G. Nuñez, A. Oberst, M. Oren, M. Overholtzer, M. Pagano, T. Panaretakis, M. Pasparakis, J.M. Penninger, D.M. Pereira, S. Pervaiz, M.E. Peter, M. Piacentini, P. Pinton, J.H. Prehn, H. Puthalakath, G.A. Rabinovich, M. Rehm, R. Rizzuto, C.M. Rodrigues, D.C. Rubinsztein, T. Rudel, K.M. Ryan, E. Sayan, L. Scorrano, F. Shao, Y. Shi, J. Silke, H. Simon, A. Sistigu, B.R. Stockwell, A. Strasser, G. Szabadkai, S.W. Tait, D. Tang, N. Tavernarakis, A. Thorburn, Y. Tsujimoto, B. Turk, T. Vanden Berghe, P. Vandenabeele, M.G. Vander Heiden, A. Villunger, H.W. Virgin, K.H. Vousden, D. Vucic, E.F. Wagner, H. Walczak, D. Wallach, Y. Wang, J.A. Wells, W. Wood, J. Yuan, Z. Zakeri, B. Zhivotovsky, L. Zitvogel, G. Melino, and G. Kroemer, "Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018", Cell Death & Differentiation, vol. 25, pp. 486-541, 2018. http://dx.doi.org/10.1038/s41418-017-0012-4
- L. Galluzzi, C. Brenner, E. Morselli, Z. Touat, and G. Kroemer, "Viral Control of Mitochondrial Apoptosis", PLoS Pathogens, vol. 4, pp. e1000018, 2008. http://dx.doi.org/10.1371/journal.ppat.1000018
- C. Martin, M. Booty, T. Rosebrock, C. Nunes-Alves, D. Desjardins, I. Keren, S. Fortune, H. Remold, and S. Behar, "Efferocytosis Is an Innate Antibacterial Mechanism", Cell Host & Microbe, vol. 12, pp. 289-300, 2012. http://dx.doi.org/10.1016/j.chom.2012.06.010
- G. van Zandbergen, M. Klinger, A. Mueller, S. Dannenberg, A. Gebert, W. Solbach, and T. Laskay, "Cutting Edge: Neutrophil Granulocyte Serves as a Vector forLeishmaniaEntry into Macrophages", The Journal of Immunology, vol. 173, pp. 6521-6525, 2004. http://dx.doi.org/10.4049/jimmunol.173.11.6521
- N.C. Peters, J.G. Egen, N. Secundino, A. Debrabant, N. Kimblin, S. Kamhawi, P. Lawyer, M.P. Fay, R.N. Germain, and D. Sacks, "In Vivo Imaging Reveals an Essential Role for Neutrophils in Leishmaniasis Transmitted by Sand Flies", Science, vol. 321, pp. 970-974, 2008. http://dx.doi.org/10.1126/science.1159194
- C.M. De Leon-Rodriguez, D.C.P. Rossi, M.S. Fu, Q. Dragotakes, C. Coelho, I. Guerrero Ros, B. Caballero, S.J. Nolan, and A. Casadevall, "The Outcome of the Cryptococcus neoformans–Macrophage Interaction Depends on Phagolysosomal Membrane Integrity", The Journal of Immunology, vol. 201, pp. 583-603, 2018. http://dx.doi.org/10.4049/jimmunol.1700958
- P. Vandenabeele, L. Galluzzi, T. Vanden Berghe, and G. Kroemer, "Molecular mechanisms of necroptosis: an ordered cellular explosion", Nature Reviews Molecular Cell Biology, vol. 11, pp. 700-714, 2010. http://dx.doi.org/10.1038/nrm2970
-
P.E. Czabotar, and J.M. Murphy, "A tale of two domains – a structural perspective of the pseudokinase,
MLKL ", The FEBS Journal, vol. 282, pp. 4268-4278, 2015. http://dx.doi.org/10.1111/febs.13504 - S.W. Lindgren, I. Stojiljkovic, and F. Heffron, "Macrophage killing is an essential virulence mechanism of Salmonella typhimurium.", Proceedings of the National Academy of Sciences, vol. 93, pp. 4197-4201, 1996. http://dx.doi.org/10.1073/pnas.93.9.4197
- N. Robinson, S. McComb, R. Mulligan, R. Dudani, L. Krishnan, and S. Sad, "Type I interferon induces necroptosis in macrophages during infection with Salmonella enterica serovar Typhimurium", Nature Immunology, vol. 13, pp. 954-962, 2012. http://dx.doi.org/10.1038/ni.2397
- T. Dallenga, U. Repnik, B. Corleis, J. Eich, R. Reimer, G.W. Griffiths, and U.E. Schaible, "M. tuberculosis-Induced Necrosis of Infected Neutrophils Promotes Bacterial Growth Following Phagocytosis by Macrophages", Cell Host & Microbe, vol. 22, pp. 519-530.e3, 2017. http://dx.doi.org/10.1016/j.chom.2017.09.003
-
J. Maelfait, L. Liverpool, A. Bridgeman, K.B. Ragan, J.W. Upton, and J. Rehwinkel, "Sensing of viral and endogenous
RNA byZBP 1/DAI induces necroptosis", The EMBO Journal, vol. 36, pp. 2529-2543, 2017. http://dx.doi.org/10.15252/embj.201796476 - J.W. Upton, and W.J. Kaiser, "DAI Another Way: Necroptotic Control of Viral Infection", Cell Host & Microbe, vol. 21, pp. 290-293, 2017. http://dx.doi.org/10.1016/j.chom.2017.01.016
- M.D. Stutz, S. Ojaimi, C. Allison, S. Preston, P. Arandjelovic, J.M. Hildebrand, J.J. Sandow, A.I. Webb, J. Silke, W.S. Alexander, and M. Pellegrini, "Necroptotic signaling is primed in Mycobacterium tuberculosis-infected macrophages, but its pathophysiological consequence in disease is restricted", Cell Death & Differentiation, vol. 25, pp. 951-965, 2017. http://dx.doi.org/10.1038/s41418-017-0031-1
- N. Kayagaki, S. Warming, M. Lamkanfi, L.V. Walle, S. Louie, J. Dong, K. Newton, Y. Qu, J. Liu, S. Heldens, J. Zhang, W.P. Lee, M. Roose-Girma, and V.M. Dixit, "Non-canonical inflammasome activation targets caspase-11", Nature, vol. 479, pp. 117-121, 2011. http://dx.doi.org/10.1038/nature10558
- C. Rogers, T. Fernandes-Alnemri, L. Mayes, D. Alnemri, G. Cingolani, and E.S. Alnemri, "Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death", Nature Communications, vol. 8, 2017. http://dx.doi.org/10.1038/ncomms14128
- Y. Wang, W. Gao, X. Shi, J. Ding, W. Liu, H. He, K. Wang, and F. Shao, "Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin", Nature, vol. 547, pp. 99-103, 2017. http://dx.doi.org/10.1038/nature22393
- T. Próchnicki, and E. Latz, "Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control", Cell Metabolism, vol. 26, pp. 71-93, 2017. http://dx.doi.org/10.1016/j.cmet.2017.06.018
- I. Jorgensen, and E.A. Miao, "Pyroptotic cell death defends against intracellular pathogens", Immunological Reviews, vol. 265, pp. 130-142, 2015. http://dx.doi.org/10.1111/imr.12287
- L. Knodler, S. Crowley, H. Sham, H. Yang, M. Wrande, C. Ma, R. Ernst, O. Steele-Mortimer, J. Celli, and B. Vallance, "Noncanonical Inflammasome Activation of Caspase-4/Caspase-11 Mediates Epithelial Defenses against Enteric Bacterial Pathogens", Cell Host & Microbe, vol. 16, pp. 249-256, 2014. http://dx.doi.org/10.1016/j.chom.2014.07.002
- A. Akhter, K. Caution, A. Abu Khweek, M. Tazi, B. Abdulrahman, D. Abdelaziz, O. Voss, A. Doseff, H. Hassan, A. Azad, L. Schlesinger, M. Wewers, M. Gavrilin, and A. Amer, "Caspase-11 Promotes the Fusion of Phagosomes Harboring Pathogenic Bacteria with Lysosomes by Modulating Actin Polymerization", Immunity, vol. 37, pp. 35-47, 2012. http://dx.doi.org/10.1016/j.immuni.2012.05.001
- N. Uwamahoro, J. Verma-Gaur, H. Shen, Y. Qu, R. Lewis, J. Lu, K. Bambery, S.L. Masters, J.E. Vince, T. Naderer, and A. Traven, "The Pathogen Candida albicans Hijacks Pyroptosis for Escape from Macrophages", mBio, vol. 5, 2014. http://dx.doi.org/10.1128/mBio.00003-14
- L. Zitvogel, O. Kepp, and G. Kroemer, "Decoding Cell Death Signals in Inflammation and Immunity", Cell, vol. 140, pp. 798-804, 2010. http://dx.doi.org/10.1016/j.cell.2010.02.015
- C.R. Collins, F. Hackett, M. Strath, M. Penzo, C. Withers-Martinez, D.A. Baker, and M.J. Blackman, "Malaria Parasite cGMP-dependent Protein Kinase Regulates Blood Stage Merozoite Secretory Organelle Discharge and Egress", PLoS Pathogens, vol. 9, pp. e1003344, 2013. http://dx.doi.org/10.1371/journal.ppat.1003344
- J.D. Dvorin, D.C. Martyn, S.D. Patel, J.S. Grimley, C.R. Collins, C.S. Hopp, A.T. Bright, S. Westenberger, E. Winzeler, M.J. Blackman, D.A. Baker, T.J. Wandless, and M.T. Duraisingh, "A Plant-Like Kinase in Plasmodium falciparum Regulates Parasite Egress from Erythrocytes", Science, vol. 328, pp. 910-912, 2010. http://dx.doi.org/10.1126/science.1188191
- S. Absalon, K. Blomqvist, R.M. Rudlaff, T.J. DeLano, M.P. Pollastri, and J.D. Dvorin, "Calcium-Dependent Protein Kinase 5 Is Required for Release of Egress-Specific Organelles in Plasmodium falciparum", mBio, vol. 9, 2018. http://dx.doi.org/10.1128/mBio.00130-18
- S. Yeoh, R.A. O'Donnell, K. Koussis, A.R. Dluzewski, K.H. Ansell, S.A. Osborne, F. Hackett, C. Withers-Martinez, G.H. Mitchell, L.H. Bannister, J.S. Bryans, C.A. Kettleborough, and M.J. Blackman, "Subcellular Discharge of a Serine Protease Mediates Release of Invasive Malaria Parasites from Host Erythrocytes", Cell, vol. 131, pp. 1072-1083, 2007. http://dx.doi.org/10.1016/j.cell.2007.10.049
- S. Arastu-Kapur, E.L. Ponder, U.P. Fonović, S. Yeoh, F. Yuan, M. Fonović, M. Grainger, C.I. Phillips, J.C. Powers, and M. Bogyo, "Identification of proteases that regulate erythrocyte rupture by the malaria parasite Plasmodium falciparum", Nature Chemical Biology, vol. 4, pp. 203-213, 2008. http://dx.doi.org/10.1038/nchembio.70
- N.C. Silmon de Monerri, H.R. Flynn, M.G. Campos, F. Hackett, K. Koussis, C. Withers-Martinez, J.M. Skehel, and M.J. Blackman, "Global Identification of Multiple Substrates forPlasmodium falciparumSUB1, an Essential Malarial Processing Protease", Infection and Immunity, vol. 79, pp. 1086-1097, 2011. http://dx.doi.org/10.1128/IAI.00902-10
- A. Ruecker, M. Shea, F. Hackett, C. Suarez, E.M. Hirst, K. Milutinovic, C. Withers-Martinez, and M.J. Blackman, "Proteolytic Activation of the Essential Parasitophorous Vacuole Cysteine Protease SERA6 Accompanies Malaria Parasite Egress from Its Host Erythrocyte", Journal of Biological Chemistry, vol. 287, pp. 37949-37963, 2012. http://dx.doi.org/10.1074/jbc.M112.400820
-
R. Stallmach, M. Kavishwar, C. Withers‐Martinez, F. Hackett, C.R. Collins, S.A. Howell, S. Yeoh, E. Knuepfer, A.J. Atid, A.A. Holder, and M.J. Blackman, "
P lasmodium falciparumSERA 5 plays a non‐enzymatic role in the malarial asexual blood‐stage lifecycle", Molecular Microbiology, vol. 96, pp. 368-387, 2015. http://dx.doi.org/10.1111/mmi.12941 - C.R. Collins, F. Hackett, J. Atid, M.S.Y. Tan, and M.J. Blackman, "The Plasmodium falciparum pseudoprotease SERA5 regulates the kinetics and efficiency of malaria parasite egress from host erythrocytes", PLOS Pathogens, vol. 13, pp. e1006453, 2017. http://dx.doi.org/10.1371/journal.ppat.1006453
- J.A. Thomas, M.S.Y. Tan, C. Bisson, A. Borg, T.R. Umrekar, F. Hackett, V.L. Hale, G. Vizcay-Barrena, R.A. Fleck, A.P. Snijders, H.R. Saibil, and M.J. Blackman, "A protease cascade regulates release of the human malaria parasite Plasmodium falciparum from host red blood cells", Nature Microbiology, vol. 3, pp. 447-455, 2018. http://dx.doi.org/10.1038/s41564-018-0111-0
- S. Glushakova, J. Mazar, M.F. Hohmann-Marriott, E. Hama, and J. Zimmerberg, "Irreversible effect of cysteine protease inhibitors on the release of malaria parasites from infected erythrocytes", Cellular Microbiology, vol. 11, pp. 95-105, 2009. http://dx.doi.org/10.1111/j.1462-5822.2008.01242.x
- S. Glushakova, G. Humphrey, E. Leikina, A. Balaban, J. Miller, and J. Zimmerberg, "New Stages in the Program of Malaria Parasite Egress Imaged in Normal and Sickle Erythrocytes", Current Biology, vol. 20, pp. 1117-1121, 2010. http://dx.doi.org/10.1016/j.cub.2010.04.051
- S. Glushakova, J.R. Beck, M. Garten, B.L. Busse, A.S. Nasamu, T. Tenkova-Heuser, J. Heuser, D.E. Goldberg, and J. Zimmerberg, "Rounding precedes rupture and breakdown of vacuolar membranes minutes before malaria parasite egress from erythrocytes", Cellular Microbiology, vol. 20, pp. e12868, 2018. http://dx.doi.org/10.1111/cmi.12868
- V.L. Hale, J.M. Watermeyer, F. Hackett, G. Vizcay-Barrena, C. van Ooij, J.A. Thomas, M.C. Spink, M. Harkiolaki, E. Duke, R.A. Fleck, M.J. Blackman, and H.R. Saibil, "Parasitophorous vacuole poration precedes its rupture and rapid host erythrocyte cytoskeleton collapse in Plasmodium falciparum egress", Proceedings of the National Academy of Sciences, vol. 114, pp. 3439-3444, 2017. http://dx.doi.org/10.1073/pnas.1619441114
- S. Glushakova, D. Yin, T. Li, and J. Zimmerberg, "Membrane Transformation during Malaria Parasite Release from Human Red Blood Cells", Current Biology, vol. 15, pp. 1645-1650, 2005. http://dx.doi.org/10.1016/j.cub.2005.07.067
- M. Abkarian, G. Massiera, L. Berry, M. Roques, and C. Braun-Breton, "A novel mechanism for egress of malarial parasites from red blood cells", Blood, vol. 117, pp. 4118-4124, 2011. http://dx.doi.org/10.1182/blood-2010-08-299883
- A.J. Perrin, C.R. Collins, M.R.G. Russell, L.M. Collinson, D.A. Baker, and M.J. Blackman, "The Actinomyosin Motor Drives Malaria Parasite Red Blood Cell Invasion but Not Egress", mBio, vol. 9, 2018. http://dx.doi.org/10.1128/mBio.00905-18
- G. PRADEL, "Proteins of the malaria parasite sexual stages: expression, function and potential for transmission blocking strategies", Parasitology, vol. 134, pp. 1911-1929, 2007. http://dx.doi.org/10.1017/s0031182007003381
- A. Kuehn, and G. Pradel, "The Coming-Out of Malaria Gametocytes", Journal of Biomedicine and Biotechnology, vol. 2010, pp. 1-11, 2010. http://dx.doi.org/10.1155/2010/976827
- S. Bennink, M.J. Kiesow, and G. Pradel, "The development of malaria parasites in the mosquito midgut", Cellular Microbiology, vol. 18, pp. 905-918, 2016. http://dx.doi.org/10.1111/cmi.12604
- D.K. Muhia, C.A. Swales, W. Deng, J.M. Kelly, and D.A. Baker, "The gametocyte‐activating factor xanthurenic acid stimulates an increase in membrane‐associated guanylyl cyclase activity in the human malaria parasite Plasmodium falciparum", Molecular Microbiology, vol. 42, pp. 553-560, 2001. http://dx.doi.org/10.1046/j.1365-2958.2001.02665.x
- L. McRobert, C.J. Taylor, W. Deng, Q.L. Fivelman, R.M. Cummings, S.D. Polley, O. Billker, and D.A. Baker, "Gametogenesis in Malaria Parasites Is Mediated by the cGMP-Dependent Protein Kinase", PLoS Biology, vol. 6, pp. e139, 2008. http://dx.doi.org/10.1371/journal.pbio.0060139
- M. Brochet, M.O. Collins, T.K. Smith, E. Thompson, S. Sebastian, K. Volkmann, F. Schwach, L. Chappell, A.R. Gomes, M. Berriman, J.C. Rayner, D.A. Baker, J. Choudhary, and O. Billker, "Phosphoinositide Metabolism Links cGMP-Dependent Protein Kinase G to Essential Ca2+ Signals at Key Decision Points in the Life Cycle of Malaria Parasites", PLoS Biology, vol. 12, pp. e1001806, 2014. http://dx.doi.org/10.1371/journal.pbio.1001806
- A.C. Raabe, K. Wengelnik, O. Billker, and H.J. Vial, "Multiple roles for Plasmodium berghei phosphoinositide-specific phospholipase C in regulating gametocyte activation and differentiation", Cellular Microbiology, vol. 13, pp. 955-966, 2011. http://dx.doi.org/10.1111/j.1462-5822.2011.01591.x
- O. Billker, S. Dechamps, R. Tewari, G. Wenig, B. Franke-Fayard, and V. Brinkmann, "Calcium and a Calcium-Dependent Protein Kinase Regulate Gamete Formation and Mosquito Transmission in a Malaria Parasite", Cell, vol. 117, pp. 503-514, 2004. http://dx.doi.org/10.1016/s0092-8674(04)00449-0
- S. Sebastian, M. Brochet, M. Collins, F. Schwach, M. Jones, D. Goulding, J. Rayner, J. Choudhary, and O. Billker, "A Plasmodium Calcium-Dependent Protein Kinase Controls Zygote Development and Transmission by Translationally Activating Repressed mRNAs", Cell Host & Microbe, vol. 12, pp. 9-19, 2012. http://dx.doi.org/10.1016/j.chom.2012.05.014
- P. Alano, D. Read, M. Bruce, M. Aikawa, T. Kaido, T. Tegoshi, S. Bhatti, D.K. Smith, C. Luo, S. Hansra, R. Carter, and J.F. Elliott, "COS cell expression cloning of Pfg377, a Plasmodium falciparum gametocyte antigen associated with osmiophilic bodies", Molecular and Biochemical Parasitology, vol. 74, pp. 143-156, 1995. http://dx.doi.org/10.1016/0166-6851(95)02491-3
- C. Severini, F. Silvestrini, A. Sannella, S. Barca, L. Gradoni, and P. Alano, "The production of the osmiophilic body protein Pfg377 is associated with stage of maturation and sex in Plasmodium falciparum gametocytes", Molecular and Biochemical Parasitology, vol. 100, pp. 247-252, 1999. http://dx.doi.org/10.1016/s0166-6851(99)00050-x
- M. Ponzi, I. Sidén-Kiamos, L. Bertuccini, C. Currà, H. Kroeze, G. Camarda, T. Pace, B. Franke-Fayard, E.C. Laurentino, C. Louis, A.P. Waters, C.J. Janse, and P. Alano, "Egress ofPlasmodium bergheigametes from their host erythrocyte is mediated by the MDV-1/PEG3 protein", Cellular Microbiology, vol. 11, pp. 1272-1288, 2009. http://dx.doi.org/10.1111/j.1462-5822.2009.01331.x
- A.M. Talman, C. Lacroix, S.R. Marques, A.M. Blagborough, R. Carzaniga, R. Ménard, and R.E. Sinden, "PbGEST mediates malaria transmission to both mosquito and vertebrate host", Molecular Microbiology, vol. 82, pp. 462-474, 2011. http://dx.doi.org/10.1111/j.1365-2958.2011.07823.x
- A. Olivieri, L. Bertuccini, E. Deligianni, B. Franke-Fayard, C. Currà, I. Siden-Kiamos, E. Hanssen, F. Grasso, F. Superti, T. Pace, F. Fratini, C.J. Janse, and M. Ponzi, "Distinct properties of the egress-related osmiophilic bodies in male and female gametocytes of the rodent malaria parasitePlasmodium berghei", Cellular Microbiology, vol. 17, pp. 355-368, 2014. http://dx.doi.org/10.1111/cmi.12370
- J. Kehrer, F. Frischknecht, and G.R. Mair, "Proteomic Analysis of the Plasmodium berghei Gametocyte Egressome and Vesicular bioID of Osmiophilic Body Proteins Identifies Merozoite TRAP-like Protein (MTRAP) as an Essential Factor for Parasite Transmission", Molecular & Cellular Proteomics, vol. 15, pp. 2852-2862, 2016. http://dx.doi.org/10.1074/mcp.M116.058263
- J. Kehrer, M. Singer, L. Lemgruber, P.A.G.C. Silva, F. Frischknecht, and G.R. Mair, "A Putative Small Solute Transporter Is Responsible for the Secretion of G377 and TRAP-Containing Secretory Vesicles during Plasmodium Gamete Egress and Sporozoite Motility", PLOS Pathogens, vol. 12, pp. e1005734, 2016. http://dx.doi.org/10.1371/journal.ppat.1005734
- P. Suárez-Cortés, V. Sharma, L. Bertuccini, G. Costa, N. Bannerman, A. Rosa Sannella, K. Williamson, M. Klemba, E.A. Levashina, E. Lasonder, and P. Alano, "Comparative Proteomics and Functional Analysis Reveal a Role of Plasmodium falciparum Osmiophilic Bodies in Malaria Parasite Transmission", Molecular & Cellular Proteomics, vol. 15, pp. 3243-3255, 2016. http://dx.doi.org/10.1074/mcp.M116.060681
- T. Weißbach, A. Golzmann, S. Bennink, G. Pradel, and C. Julius Ngwa, "Transcript and protein expression analysis of proteases in the blood stages of Plasmodium falciparum", Experimental Parasitology, vol. 180, pp. 33-44, 2017. http://dx.doi.org/10.1016/j.exppara.2017.03.006
- D.Y. Bargieri, S. Thiberge, C.L. Tay, A.F. Carey, A. Rantz, F. Hischen, A. Lorthiois, U. Straschil, P. Singh, S. Singh, T. Triglia, T. Tsuboi, A. Cowman, C. Chitnis, P. Alano, J. Baum, G. Pradel, C. Lavazec, and R. Ménard, "Plasmodium Merozoite TRAP Family Protein Is Essential for Vacuole Membrane Disruption and Gamete Egress from Erythrocytes", Cell Host & Microbe, vol. 20, pp. 618-630, 2016. http://dx.doi.org/10.1016/j.chom.2016.10.015
- J. Baum, D. Richard, J. Healer, M. Rug, Z. Krnajski, T. Gilberger, J.L. Green, A.A. Holder, and A.F. Cowman, "A Conserved Molecular Motor Drives Cell Invasion and Gliding Motility across Malaria Life Cycle Stages and Other Apicomplexan Parasites", Journal of Biological Chemistry, vol. 281, pp. 5197-5208, 2006. http://dx.doi.org/10.1074/jbc.M509807200
- E. Deligianni, R.N. Morgan, L. Bertuccini, C.C. Wirth, N.C. Silmon de Monerri, L. Spanos, M.J. Blackman, C. Louis, G. Pradel, and I. Siden-Kiamos, "A perforin-like protein mediates disruption of the erythrocyte membrane during egress ofPlasmodium bergheimale gametocytes", Cellular Microbiology, vol. 15, pp. 1438-1455, 2013. http://dx.doi.org/10.1111/cmi.12131
-
C.C. Wirth, S. Glushakova, M. Scheuermayer, U. Repnik, S. Garg, D. Schaack, M.M. Kachman, T. Weißbach, J. Zimmerberg, T. Dandekar, G. Griffiths, C.E. Chitnis, S. Singh, R. Fischer, and G. Pradel, "Perforin‐like protein
PPLP 2 permeabilizes the red blood cell membrane during egress ofP lasmodium falciparum gametocytes", Cellular Microbiology, vol. 16, pp. 709-733, 2014. http://dx.doi.org/10.1111/cmi.12288 - L. Sologub, A. Kuehn, S. Kern, J. Przyborski, R. Schillig, and G. Pradel, "Malaria proteases mediate inside-out egress of gametocytes from red blood cells following parasite transmission to the mosquito", Cellular Microbiology, vol. 13, pp. 897-912, 2011. http://dx.doi.org/10.1111/j.1462-5822.2011.01588.x
- M. Andreadaki, E. Hanssen, E. Deligianni, C. Claudet, K. Wengelnik, V. Mollard, G.I. McFadden, M. Abkarian, C. Braun-Breton, and I. Siden-Kiamos, "Sequential Membrane Rupture and Vesiculation during Plasmodium berghei Gametocyte Egress from the Red Blood Cell", Scientific Reports, vol. 8, 2018. http://dx.doi.org/10.1038/s41598-018-21801-3
- I. Rupp, R. Bosse, T. Schirmeister, and G. Pradel, "Effect of protease inhibitors on exflagellation in Plasmodium falciparum", Molecular and Biochemical Parasitology, vol. 158, pp. 208-212, 2008. http://dx.doi.org/10.1016/j.molbiopara.2007.12.009
- V. Pszenny, K. Ehrenman, J.D. Romano, A. Kennard, A. Schultz, D.S. Roos, M.E. Grigg, V.B. Carruthers, and I. Coppens, "A Lipolytic Lecithin:Cholesterol Acyltransferase Secreted by Toxoplasma Facilitates Parasite Replication and Egress", Journal of Biological Chemistry, vol. 291, pp. 3725-3746, 2016. http://dx.doi.org/10.1074/jbc.M115.671974
- A.J. Schultz, and V.B. Carruthers, "Toxoplasma gondii LCAT Primarily Contributes to Tachyzoite Egress", mSphere, vol. 3, 2018. http://dx.doi.org/10.1128/mSphereDirect.00073-18
- B.F.C. Kafsack, J.D.O. Pena, I. Coppens, S. Ravindran, J.C. Boothroyd, and V.B. Carruthers, "Rapid Membrane Disruption by a Perforin-Like Protein Facilitates Parasite Exit from Host Cells", Science, vol. 323, pp. 530-533, 2009. http://dx.doi.org/10.1126/science.1165740
- M.S. Roiko, and V.B. Carruthers, "Functional Dissection of Toxoplasma gondii Perforin-like Protein 1 Reveals a Dual Domain Mode of Membrane Binding for Cytolysis and Parasite Egress", Journal of Biological Chemistry, vol. 288, pp. 8712-8725, 2013. http://dx.doi.org/10.1074/jbc.M113.450932
- T. Ni, S.I. Williams, S. Rezelj, G. Anderluh, K. Harlos, P.J. Stansfeld, and R.J.C. Gilbert, "Structures of monomeric and oligomeric forms of the Toxoplasma gondii perforin-like protein 1", Science Advances, vol. 4, 2018. http://dx.doi.org/10.1126/sciadv.aaq0762
- S.K. Dogga, B. Mukherjee, D. Jacot, T. Kockmann, L. Molino, P. Hammoudi, R.C. Hartkoorn, A.B. Hehl, and D. Soldati-Favre, "A druggable secretory protein maturase of Toxoplasma essential for invasion and egress", eLife, vol. 6, 2017. http://dx.doi.org/10.7554/eLife.27480
- S. Gras, A. Jackson, S. Woods, G. Pall, J. Whitelaw, J.M. Leung, G.E. Ward, C.W. Roberts, and M. Meissner, "Parasites lacking the micronemal protein MIC2 are deficient in surface attachment and host cell egress, but remain virulent in vivo", Wellcome Open Research, vol. 2, pp. 32, 2017. http://dx.doi.org/10.12688/wellcomeopenres.11594.2
- S. Saha, B.I. Coleman, R. Dubey, I.J. Blader, and M. Gubbels, "Two Phosphoglucomutase Paralogs Facilitate Ionophore-Triggered Secretion of theToxoplasmaMicronemes", mSphere, vol. 2, 2017. http://dx.doi.org/10.1128/mSphere.00521-17
- R. Moudy, T.J. Manning, and C.J. Beckers, "The Loss of Cytoplasmic Potassium upon Host Cell Breakdown Triggers Egress of Toxoplasma gondii", Journal of Biological Chemistry, vol. 276, pp. 41492-41501, 2001. http://dx.doi.org/10.1074/jbc.M106154200
- M.S. Roiko, N. Svezhova, and V.B. Carruthers, "Acidification Activates Toxoplasma gondii Motility and Egress by Enhancing Protein Secretion and Cytolytic Activity", PLoS Pathogens, vol. 10, pp. e1004488, 2014. http://dx.doi.org/10.1371/journal.ppat.1004488
- K.M. Brown, S. Lourido, and L.D. Sibley, "Serum Albumin Stimulates Protein Kinase G-dependent Microneme Secretion in Toxoplasma gondii", Journal of Biological Chemistry, vol. 291, pp. 9554-9565, 2016. http://dx.doi.org/10.1074/jbc.M115.700518
- R.J. Stewart, L. Whitehead, B. Nijagal, B.E. Sleebs, G. Lessene, M.J. McConville, K.L. Rogers, and C.J. Tonkin, "Analysis of Ca 2 + mediated signaling regulating Toxoplasma infectivity reveals complex relationships between key molecules", Cellular Microbiology, vol. 19, 2017. http://dx.doi.org/10.1111/cmi.12685
- E. Chini, K. Nagamune, D. Wetzel, and L. Sibley, "Evidence that the cADPR signalling pathway controls calcium-mediated microneme secretion in Toxoplasma gondii", Biochemical Journal, vol. 389, pp. 269-277, 2005. http://dx.doi.org/10.1042/BJ20041971
-
Y. Jia, J. Marq, H. Bisio, D. Jacot, C. Mueller, L. Yu, J. Choudhary, M. Brochet, and D. Soldati‐Favre, "Crosstalk between
PKA andPKG controlspH ‐dependent host cell egress of Toxoplasma gondii", The EMBO Journal, vol. 36, pp. 3250-3267, 2017. http://dx.doi.org/10.15252/embj.201796794 - K. Nagamune, L.M. Hicks, B. Fux, F. Brossier, E.N. Chini, and L.D. Sibley, "Abscisic acid controls calcium-dependent egress and development in Toxoplasma gondii", Nature, vol. 451, pp. 207-210, 2008. http://dx.doi.org/10.1038/nature06478
- R.D. Arroyo-Olarte, J.F. Brouwers, A. Kuchipudi, J.B. Helms, A. Biswas, I.R. Dunay, R. Lucius, and N. Gupta, "Phosphatidylthreonine and Lipid-Mediated Control of Parasite Virulence", PLOS Biology, vol. 13, pp. e1002288, 2015. http://dx.doi.org/10.1371/journal.pbio.1002288
- A. Kuchipudi, R.D. Arroyo-Olarte, F. Hoffmann, V. Brinkmann, and N. Gupta, "Optogenetic monitoring identifies phosphatidylthreonine-regulated calcium homeostasis in Toxoplasma gondii", Microbial Cell, vol. 3, pp. 215-223, 2016. http://dx.doi.org/10.15698/mic2016.05.500
- P. Kong, C. Ufermann, D.L. Zimmermann, Q. Yin, X. Suo, J.B. Helms, J.F. Brouwers, and N. Gupta, "Two phylogenetically and compartmentally distinct CDP-diacylglycerol synthases cooperate for lipid biogenesis in Toxoplasma gondii", Journal of Biological Chemistry, vol. 292, pp. 7145-7159, 2017. http://dx.doi.org/10.1074/jbc.M116.765487
- S. Lourido, J. Shuman, C. Zhang, K.M. Shokat, R. Hui, and L.D. Sibley, "Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma", Nature, vol. 465, pp. 359-362, 2010. http://dx.doi.org/10.1038/nature09022
- J.M. McCoy, R.J. Stewart, A.D. Uboldi, D. Li, J. Schröder, N.E. Scott, A.T. Papenfuss, A.M. Lehane, L.J. Foster, and C.J. Tonkin, "A forward genetic screen identifies a negative regulator of rapid Ca2+-dependent cell egress (MS1) in the intracellular parasite Toxoplasma gondii", Journal of Biological Chemistry, vol. 292, pp. 7662-7674, 2017. http://dx.doi.org/10.1074/jbc.M117.775114
- J.M. McCoy, L. Whitehead, G.G. van Dooren, and C.J. Tonkin, "TgCDPK3 Regulates Calcium-Dependent Egress of Toxoplasma gondii from Host Cells", PLoS Pathogens, vol. 8, pp. e1003066, 2012. http://dx.doi.org/10.1371/journal.ppat.1003066
- R.Y. Gaji, D.E. Johnson, M. Treeck, M. Wang, A. Hudmon, and G. Arrizabalaga, "Phosphorylation of a Myosin Motor by TgCDPK3 Facilitates Rapid Initiation of Motility during Toxoplasma gondii egress", PLOS Pathogens, vol. 11, pp. e1005268, 2015. http://dx.doi.org/10.1371/journal.ppat.1005268
- H. Bullen, Y. Jia, Y. Yamaryo-Botté, H. Bisio, O. Zhang, N. Jemelin, J. Marq, V. Carruthers, C. Botté, and D. Soldati-Favre, "Phosphatidic Acid-Mediated Signaling Regulates Microneme Secretion in Toxoplasma", Cell Host & Microbe, vol. 19, pp. 349-360, 2016. http://dx.doi.org/10.1016/j.chom.2016.02.006
- P. Burda, M.A. Roelli, M. Schaffner, S.M. Khan, C.J. Janse, and V.T. Heussler, "A Plasmodium Phospholipase Is Involved in Disruption of the Liver Stage Parasitophorous Vacuole Membrane", PLOS Pathogens, vol. 11, pp. e1004760, 2015. http://dx.doi.org/10.1371/journal.ppat.1004760
- V. Risco-Castillo, S. Topçu, C. Marinach, G. Manzoni, A. Bigorgne, S. Briquet, X. Baudin, M. Lebrun, J. Dubremetz, and O. Silvie, "Malaria Sporozoites Traverse Host Cells within Transient Vacuoles", Cell Host & Microbe, vol. 18, pp. 593-603, 2015. http://dx.doi.org/10.1016/j.chom.2015.10.006
- A.S. Yang, M.T. O’Neill, C. Jennison, S. Lopaticki, C.C. Allison, J.S. Armistead, S.M. Erickson, K.L. Rogers, A.M. Ellisdon, J.C. Whisstock, R.E. Tweedell, R.R. Dinglasan, D.N. Douglas, N.M. Kneteman, and J.A. Boddey, "Cell Traversal Activity Is Important for Plasmodium falciparum Liver Infection in Humanized Mice", Cell Reports, vol. 18, pp. 3105-3116, 2017. http://dx.doi.org/10.1016/j.celrep.2017.03.017
- F.S.M. Noronha, J.S. Cruz, P.S.L. Beirão, and M.F. Horta, "Macrophage Damage byLeishmania amazonensisCytolysin: Evidence of Pore Formation on Cell Membrane", Infection and Immunity, vol. 68, pp. 4578-4584, 2000. http://dx.doi.org/10.1128/iai.68.8.4578-4584.2000
- T. Castro-Gomes, F.R. Almeida-Campos, C.E. Calzavara-Silva, R.A. da Silva, F. Frézard, and M.F. Horta, "Membrane binding requirements for the cytolytic activity of Leishmania amazonensis leishporin", FEBS Letters, vol. 583, pp. 3209-3214, 2009. http://dx.doi.org/10.1016/j.febslet.2009.09.005
- F.R. Almeida-Campos, T. Castro-Gomes, A. Machado-Silva, J.S. de Oliveira, M.M. Santoro, F. Frézard, and M.F. Horta, "Activation of Leishmania spp. leishporin: evidence that dissociation of an inhibitor not only improves its lipid-binding efficiency but also endows it with the ability to form pores", Parasitology Research, vol. 112, pp. 3305-3314, 2013. http://dx.doi.org/10.1007/s00436-013-3510-4
- S.S.C. Rubin-de-Celis, H. Uemura, N. Yoshida, and S. Schenkman, "Expression of trypomastigote trans-sialidase in metacyclic forms of Trypanosoma cruzi increases parasite escape from its parasitophorous vacuole", Cellular Microbiology, vol. 8, pp. 1888-1898, 2006. http://dx.doi.org/10.1111/j.1462-5822.2006.00755.x
- L. Freire-de-Lima, L.M.D. Fonseca, V.A. da Silva, K.M. da Costa, A. Morrot, C.G. Freire-de-Lima, J.O. Previato, and L. Mendonça-Previato, "Modulation of Cell Sialoglycophenotype: A Stylish Mechanism Adopted by Trypanosoma cruzi to Ensure Its Persistence in the Infected Host", Frontiers in Microbiology, vol. 7, 2016. http://dx.doi.org/10.3389/fmicb.2016.00698
- N.W. Andrews, C.K. Abrams, S.L. Slatin, and G. Griffiths, "A T. cruzi-secreted protein immunologically related to the complement component C9: Evidence for membrane pore-forming activity at low pH", Cell, vol. 61, pp. 1277-1287, 1990. http://dx.doi.org/10.1016/0092-8674(90)90692-8
- B.F. Hall, P. Webster, A.K. Ma, K.A. Joiner, and N.W. Andrews, "Desialylation of lysosomal membrane glycoproteins by Trypanosoma cruzi: a role for the surface neuraminidase in facilitating parasite entry into the host cell cytoplasm.", The Journal of experimental medicine, vol. 176, pp. 313-325, 1992. http://dx.doi.org/10.1084/jem.176.2.313
- E.D.R. Case, and J.E. Samuel, "Contrasting Lifestyles Within the Host Cell", Microbiology Spectrum, vol. 4, 2016. http://dx.doi.org/10.1128/microbiolspec.vmbf-0014-2015
- D.A. Portnoy, P.S. Jacks, and D.J. Hinrichs, "Role of hemolysin for the intracellular growth of Listeria monocytogenes.", The Journal of experimental medicine, vol. 167, pp. 1459-1471, 1988. http://dx.doi.org/10.1084/jem.167.4.1459
- L.W. Cheng, and D.A. Portnoy, "Drosophila S2 cells: an alternative infection model for Listeria monocytogenes", Cellular Microbiology, vol. 5, pp. 875-885, 2003. http://dx.doi.org/10.1046/j.1462-5822.2003.00327.x
- C.L. Birmingham, V. Canadien, N.A. Kaniuk, B.E. Steinberg, D.E. Higgins, and J.H. Brumell, "Listeriolysin O allows Listeria monocytogenes replication in macrophage vacuoles", Nature, vol. 451, pp. 350-354, 2008. http://dx.doi.org/10.1038/nature06479
- L.G. Tilney, and D.A. Portnoy, "Actin filaments and the growth, movement, and spread of the intracellular bacterial parasite, Listeria monocytogenes.", The Journal of cell biology, vol. 109, pp. 1597-1608, 1989. http://dx.doi.org/10.1083/jcb.109.4.1597
- J.L. Gaillard, P. Berche, J. Mounier, S. Richard, and P. Sansonetti, "In vitro model of penetration and intracellular growth of Listeria monocytogenes in the human enterocyte-like cell line Caco-2.", Infection and immunity, 1987. http://www.ncbi.nlm.nih.gov/pubmed/3117693
- A. Camilli, L.G. Tilney, and D.A. Portnoy, "Dual roles of plcA in Listeria monocytogenes pathogenesis", Molecular Microbiology, vol. 8, pp. 143-157, 1993. http://dx.doi.org/10.1111/j.1365-2958.1993.tb01211.x
- G.A. Smith, H. Marquis, S. Jones, N.C. Johnston, D.A. Portnoy, and H. Goldfine, "The two distinct phospholipases C of Listeria monocytogenes have overlapping roles in escape from a vacuole and cell-to-cell spread.", Infection and immunity, 1995. http://www.ncbi.nlm.nih.gov/pubmed/7591052
- S. Seveau, "Multifaceted Activity of Listeriolysin O, the Cholesterol-Dependent Cytolysin of Listeria monocytogenes", MACPF/CDC Proteins - Agents of Defence, Attack and Invasion, pp. 161-195, 2014. http://dx.doi.org/10.1007/978-94-017-8881-6_9
- I. Tattoli, M.T. Sorbara, C. Yang, S.A. Tooze, D.J. Philpott, and S.E. Girardin, "Listeriaphospholipases subvert host autophagic defenses by stalling pre-autophagosomal structures", The EMBO Journal, vol. 32, pp. 3066-3078, 2013. http://dx.doi.org/10.1038/emboj.2013.234
- G. Mitchell, L. Ge, Q. Huang, C. Chen, S. Kianian, M.F. Roberts, R. Schekman, and D.A. Portnoy, "Avoidance of Autophagy Mediated by PlcA or ActA Is Required for Listeria monocytogenes Growth in Macrophages", Infection and Immunity, vol. 83, pp. 2175-2184, 2015. http://dx.doi.org/10.1128/IAI.00110-15
- K.E. Beauregard, K. Lee, R.J. Collier, and J.A. Swanson, "pH-dependent Perforation of Macrophage Phagosomes by Listeriolysin O from Listeria monocytogenes ", The Journal of Experimental Medicine, vol. 186, pp. 1159-1163, 1997. http://dx.doi.org/10.1084/jem.186.7.1159
- I.J. Glomski, M.M. Gedde, A.W. Tsang, J.A. Swanson, and D.A. Portnoy, "The Listeria monocytogenes hemolysin has an acidic pH optimum to compartmentalize activity and prevent damage to infected host cells", The Journal of Cell Biology, vol. 156, pp. 1029-1038, 2002. http://dx.doi.org/10.1083/jcb.200201081
- L.M. Shaughnessy, A.D. Hoppe, K.A. Christensen, and J.A. Swanson, "Membrane perforations inhibit lysosome fusion by altering pH and calcium in Listeria monocytogenes vacuoles", Cellular Microbiology, vol. 8, pp. 781-792, 2006. http://dx.doi.org/10.1111/j.1462-5822.2005.00665.x
- N. High, J. Mounier, M. Prévost, and P. Sansonetti, "IpaB of Shigella flexneri causes entry into epithelial cells and escape from the phagocytic vacuole.", The EMBO Journal, vol. 11, pp. 1991-1999, 1992. http://dx.doi.org/10.1002/j.1460-2075.1992.tb05253.x
- A. Page, H. Ohayon, P.J. Sansonetti, and C. Parsot, "The secreted IpaB and IpaC invasins and their cytoplasmic chaperone IpgC are required for intercellular dissemination of Shigella flexneri", Cellular Microbiology, vol. 1, pp. 183-193, 1999. http://dx.doi.org/10.1046/j.1462-5822.1999.00019.x
- R.D. Hayward, R.J. Cain, E.J. McGhie, N. Phillips, M.J. Garner, and V. Koronakis, "Cholesterol binding by the bacterial type III translocon is essential for virulence effector delivery into mammalian cells", Molecular Microbiology, vol. 56, pp. 590-603, 2005. http://dx.doi.org/10.1111/j.1365-2958.2005.04568.x
- W.L. Picking, H. Nishioka, P.D. Hearn, M.A. Baxter, A.T. Harrington, A. Blocker, and W.D. Picking, "IpaD of Shigella flexneri Is Independently Required for Regulation of Ipa Protein Secretion and Efficient Insertion of IpaB and IpaC into Host Membranes", Infection and Immunity, vol. 73, pp. 1432-1440, 2005. http://dx.doi.org/10.1128/IAI.73.3.1432-1440.2005
- L.M. Stamm, J.H. Morisaki, L. Gao, R.L. Jeng, K.L. McDonald, R. Roth, S. Takeshita, J. Heuser, M.D. Welch, and E.J. Brown, "Mycobacterium marinum Escapes from Phagosomes and Is Propelled by Actin-based Motility", The Journal of Experimental Medicine, vol. 198, pp. 1361-1368, 2003. http://dx.doi.org/10.1084/jem.20031072
- N. van der Wel, D. Hava, D. Houben, D. Fluitsma, M. van Zon, J. Pierson, M. Brenner, and P.J. Peters, "M. tuberculosis and M. leprae Translocate from the Phagolysosome to the Cytosol in Myeloid Cells", Cell, vol. 129, pp. 1287-1298, 2007. http://dx.doi.org/10.1016/j.cell.2007.05.059
- L. Gao, S. Guo, B. McLaughlin, H. Morisaki, J.N. Engel, and E.J. Brown, "A mycobacterial virulence gene cluster extending RD1 is required for cytolysis, bacterial spreading and ESAT‐6 secretion", Molecular Microbiology, vol. 53, pp. 1677-1693, 2004. http://dx.doi.org/10.1111/j.1365-2958.2004.04261.x
- M.I. de Jonge, G. Pehau-Arnaudet, M.M. Fretz, F. Romain, D. Bottai, P. Brodin, N. Honoré, G. Marchal, W. Jiskoot, P. England, S.T. Cole, and R. Brosch, "ESAT-6 fromMycobacterium tuberculosisDissociates from Its Putative Chaperone CFP-10 under Acidic Conditions and Exhibits Membrane-Lysing Activity", Journal of Bacteriology, vol. 189, pp. 6028-6034, 2007. http://dx.doi.org/10.1128/JB.00469-07
- D. Houben, C. Demangel, J. van Ingen, J. Perez, L. Baldeón, A.M. Abdallah, L. Caleechurn, D. Bottai, M. van Zon, K. de Punder, T. van der Laan, A. Kant, R. Bossers‐de Vries, P. Willemsen, W. Bitter, D. van Soolingen, R. Brosch, N. van der Wel, and P.J. Peters, "ESX‐1‐mediated translocation to the cytosol controls virulence of mycobacteria", Cellular Microbiology, vol. 14, pp. 1287-1298, 2012. http://dx.doi.org/10.1111/j.1462-5822.2012.01799.x
- J. De Leon, G. Jiang, Y. Ma, E. Rubin, S. Fortune, and J. Sun, "Mycobacterium tuberculosis ESAT-6 Exhibits a Unique Membrane-interacting Activity That Is Not Found in Its Ortholog from Non-pathogenic Mycobacterium smegmatis", Journal of Biological Chemistry, vol. 287, pp. 44184-44191, 2012. http://dx.doi.org/10.1074/jbc.M112.420869
- X. Peng, G. Jiang, W. Liu, Q. Zhang, W. Qian, and J. Sun, "Characterization of differential pore‐forming activities of ESAT‐6 proteins from Mycobacterium tuberculosis and Mycobacterium smegmatis", FEBS Letters, vol. 590, pp. 509-519, 2016. http://dx.doi.org/10.1002/1873-3468.12072
- R. Simeone, D. Bottai, W. Frigui, L. Majlessi, and R. Brosch, "ESX/type VII secretion systems of mycobacteria: Insights into evolution, pathogenicity and protection", Tuberculosis, vol. 95, pp. S150-S154, 2015. http://dx.doi.org/10.1016/j.tube.2015.02.019
- R. Simeone, L. Majlessi, J. Enninga, and R. Brosch, "Perspectives on mycobacterial vacuole-to-cytosol translocation: the importance of cytosolic access", Cellular Microbiology, vol. 18, pp. 1070-1077, 2016. http://dx.doi.org/10.1111/cmi.12622
- R. Simeone, A. Bobard, J. Lippmann, W. Bitter, L. Majlessi, R. Brosch, and J. Enninga, "Phagosomal Rupture by Mycobacterium tuberculosis Results in Toxicity and Host Cell Death", PLoS Pathogens, vol. 8, pp. e1002507, 2012. http://dx.doi.org/10.1371/journal.ppat.1002507
- R. Simeone, F. Sayes, O. Song, M.I. Gröschel, P. Brodin, R. Brosch, and L. Majlessi, "Cytosolic Access of Mycobacterium tuberculosis: Critical Impact of Phagosomal Acidification Control and Demonstration of Occurrence In Vivo", PLOS Pathogens, vol. 11, pp. e1004650, 2015. http://dx.doi.org/10.1371/journal.ppat.1004650
- G. Zhong, P. Fan, H. Ji, F. Dong, and Y. Huang, "Identification of a Chlamydial Protease–Like Activity Factor Responsible for the Degradation of Host Transcription Factors", The Journal of Experimental Medicine, vol. 193, pp. 935-942, 2001. http://dx.doi.org/10.1084/jem.193.8.935
- P.M. Bavoil, and G.I. Byrne, "Analysis of CPAF mutants: new functions, new questions (The ins and outs of a chlamydial protease)", Pathogens and Disease, vol. 71, pp. 287-291, 2014. http://dx.doi.org/10.1111/2049-632X.12194
- C. Yang, T. Starr, L. Song, J.H. Carlson, G.L. Sturdevant, P.A. Beare, W.M. Whitmire, and H.D. Caldwell, "Chlamydial Lytic Exit from Host Cells Is Plasmid Regulated", mBio, vol. 6, 2015. http://dx.doi.org/10.1128/mBio.01648-15
- K. Hybiske, and R.S. Stephens, "Mechanisms of host cell exit by the intracellular bacteriumChlamydia", Proceedings of the National Academy of Sciences, vol. 104, pp. 11430-11435, 2007. http://dx.doi.org/10.1073/pnas.0703218104
- O. Steele-Mortimer, S. Meresse, J. Gorvel, B. Toh, and B.B. Finlay, "Biogenesis of Salmonella typhimurium-containing vacuoles in epithelial cells involves interactions with the early endocytic pathway", Cellular Microbiology, vol. 1, pp. 33-49, 1999. http://dx.doi.org/10.1046/j.1462-5822.1999.00003.x
- Y. Zhang, W. Higashide, S. Dai, D.M. Sherman, and D. Zhou, "Recognition and Ubiquitination of Salmonella Type III Effector SopA by a Ubiquitin E3 Ligase, HsRMA1", Journal of Biological Chemistry, vol. 280, pp. 38682-38688, 2005. http://dx.doi.org/10.1074/jbc.M506309200
- D. Drecktrah, L.A. Knodler, D. Howe, and O. Steele‐Mortimer, "Salmonella Trafficking is Defined by Continuous Dynamic Interactions with the Endolysosomal System", Traffic, vol. 8, pp. 212-225, 2006. http://dx.doi.org/10.1111/j.1600-0854.2006.00529.x
- M.A. Bakowski, V. Braun, and J.H. Brumell, "Salmonella‐Containing Vacuoles: Directing Traffic and Nesting to Grow", Traffic, vol. 9, pp. 2022-2031, 2008. http://dx.doi.org/10.1111/j.1600-0854.2008.00827.x
- L.A. Knodler, "Salmonella enterica: living a double life in epithelial cells", Current Opinion in Microbiology, vol. 23, pp. 23-31, 2015. http://dx.doi.org/10.1016/j.mib.2014.10.010
- L.A. Knodler, B.A. Vallance, J. Celli, S. Winfree, B. Hansen, M. Montero, and O. Steele-Mortimer, "Dissemination of invasiveSalmonellavia bacterial-induced extrusion of mucosal epithelia", Proceedings of the National Academy of Sciences, vol. 107, pp. 17733-17738, 2010. http://dx.doi.org/10.1073/pnas.1006098107
- T. Miki, N. Okada, Y. Shimada, and H. Danbara, "Characterization of Salmonella pathogenicity island 1 type III secretion-dependent hemolytic activity in Salmonella enterica serovar Typhimurium", Microbial Pathogenesis, vol. 37, pp. 65-72, 2004. http://dx.doi.org/10.1016/j.micpath.2004.04.006
- D. Roy, D.R. Liston, V.J. Idone, A. Di, D.J. Nelson, C. Pujol, J.B. Bliska, S. Chakrabarti, and N.W. Andrews, "A Process for Controlling Intracellular Bacterial Infections Induced by Membrane Injury", Science, vol. 304, pp. 1515-1518, 2004. http://dx.doi.org/10.1126/science.1098371
- C.L. Birmingham, A.C. Smith, M.A. Bakowski, T. Yoshimori, and J.H. Brumell, "Autophagy Controls Salmonella Infection in Response to Damage to the Salmonella-containing Vacuole", Journal of Biological Chemistry, vol. 281, pp. 11374-11383, 2006. http://dx.doi.org/10.1074/jbc.M509157200
- H.B. Yu, M.A. Croxen, A.M. Marchiando, R.B.R. Ferreira, K. Cadwell, L.J. Foster, and B.B. Finlay, "Autophagy Facilitates Salmonella Replication in HeLa Cells", mBio, vol. 5, 2014. http://dx.doi.org/10.1128/mBio.00865-14
- C.R. Beuzon, "Salmonella maintains the integrity of its intracellular vacuole through the action of SifA", The EMBO Journal, vol. 19, pp. 3235-3249, 2000. http://dx.doi.org/10.1093/emboj/19.13.3235
- J. Ruiz‐Albert, X. Yu, C.R. Beuzón, A.N. Blakey, E.E. Galyov, and D.W. Holden, "Complementary activities of SseJ and SifA regulate dynamics of the Salmonella typhimurium vacuolar membrane", Molecular Microbiology, vol. 44, pp. 645-661, 2002. http://dx.doi.org/10.1046/j.1365-2958.2002.02912.x
- N.S. Lossi, N. Rolhion, A.I. Magee, C. Boyle, and D.W. Holden, "The Salmonella SPI-2 effector SseJ exhibits eukaryotic activator-dependent phospholipase A and glycerophospholipid : cholesterol acyltransferase activity", Microbiology, vol. 154, pp. 2680-2688, 2008. http://dx.doi.org/10.1099/mic.0.2008/019075-0
- E. Boucrot, T. Henry, J. Borg, J. Gorvel, and S. Méresse, "The Intracellular Fate of Salmonella Depends on the Recruitment of Kinesin", Science, vol. 308, pp. 1174-1178, 2005. http://dx.doi.org/10.1126/science.1110225
- T. Henry, C. Couillault, P. Rockenfeller, E. Boucrot, A. Dumont, N. Schroeder, A. Hermant, L.A. Knodler, P. Lecine, O. Steele-Mortimer, J. Borg, J. Gorvel, and S. Méresse, "The Salmonella effector protein PipB2 is a linker for kinesin-1", Proceedings of the National Academy of Sciences, vol. 103, pp. 13497-13502, 2006. http://dx.doi.org/10.1073/pnas.0605443103
- A. Dumont, E. Boucrot, S. Drevensek, V. Daire, J. Gorvel, C. Poüs, D.W. Holden, and S. Méresse, "SKIP, the Host Target of the Salmonella Virulence Factor SifA, Promotes Kinesin-1-Dependent Vacuolar Membrane Exchanges", Traffic, vol. 11, pp. 899-911, 2010. http://dx.doi.org/10.1111/j.1600-0854.2010.01069.x
- H.J. Newton, D.K.Y. Ang, I.R. van Driel, and E.L. Hartland, "Molecular Pathogenesis of Infections Caused byLegionella pneumophila", Clinical Microbiology Reviews, vol. 23, pp. 274-298, 2010. http://dx.doi.org/10.1128/CMR.00052-09
- J.E. Kirby, J.P. Vogel, H.L. Andrews, and R.R. Isberg, "Evidence for pore‐forming ability by Legionella pneumophila", Molecular Microbiology, vol. 27, pp. 323-336, 1998. http://dx.doi.org/10.1046/j.1365-2958.1998.00680.x
- M. Molmeret, O.A.T. Alli, M. Radulic, M. Susa, M. Doric, and Y.A. Kwaik, "The C‐terminus of IcmT is essential for pore formation and for intracellular trafficking of Legionella pneumophila within Acanthamoeba polyphaga", Molecular Microbiology, vol. 43, pp. 1139-1150, 2002. http://dx.doi.org/10.1046/j.1365-2958.2002.02842.x
- M. Molmeret, D.M. Bitar, L. Han, and Y.A. Kwaik, "Disruption of the Phagosomal Membrane and Egress ofLegionella pneumophilainto the Cytoplasm during the Last Stages of Intracellular Infection of Macrophages andAcanthamoeba polyphaga", Infection and Immunity, vol. 72, pp. 4040-4051, 2004. http://dx.doi.org/10.1128/iai.72.7.4040-4051.2004
- T.N. Silveira, and D.S. Zamboni, "Pore Formation Triggered byLegionellaspp. Is an Nlrc4 Inflammasome-Dependent Host Cell Response That Precedes Pyroptosis", Infection and Immunity, vol. 78, pp. 1403-1413, 2010. http://dx.doi.org/10.1128/IAI.00905-09
- M. Hiller, C. Lang, W. Michel, and A. Flieger, "Secreted phospholipases of the lung pathogen Legionella pneumophila", International Journal of Medical Microbiology, vol. 308, pp. 168-175, 2018. http://dx.doi.org/10.1016/j.ijmm.2017.10.002
- E.A. Creasey, and R.R. Isberg, "The protein SdhA maintains the integrity of the Legionella -containing vacuole", Proceedings of the National Academy of Sciences, vol. 109, pp. 3481-3486, 2012. http://dx.doi.org/10.1073/pnas.1121286109
- C. Lang, M. Hiller, and A. Flieger, "Disulfide loop cleavage of Legionella pneumophila PlaA boosts lysophospholipase A activity", Scientific Reports, vol. 7, 2017. http://dx.doi.org/10.1038/s41598-017-12796-4
- A.S. Aly, and K. Matuschewski, "A malarial cysteine protease is necessary for Plasmodium sporozoite egress from oocysts", The Journal of Experimental Medicine, vol. 202, pp. 225-230, 2005. http://dx.doi.org/10.1084/jem.20050545
- B.S. Mastan, S.K. Narwal, S. Dey, K.A. Kumar, and S. Mishra, "Plasmodium berghei plasmepsin VIII is essential for sporozoite gliding motility", International Journal for Parasitology, vol. 47, pp. 239-245, 2017. http://dx.doi.org/10.1016/j.ijpara.2016.11.009
- G. Pradel, K. Hayton, L. Aravind, L.M. Iyer, M.S. Abrahamsen, A. Bonawitz, C. Mejia, and T.J. Templeton, "A Multidomain Adhesion Protein Family Expressed inPlasmodium falciparumIs Essential for Transmission to the Mosquito", The Journal of Experimental Medicine, vol. 199, pp. 1533-1544, 2004. http://dx.doi.org/10.1084/jem.20031274
- J.D. Raine, A. Ecker, J. Mendoza, R. Tewari, R.R. Stanway, and R.E. Sinden, "Female Inheritance of Malarial lap Genes Is Essential for Mosquito Transmission", PLoS Pathogens, vol. 3, pp. e30, 2007. http://dx.doi.org/10.1371/journal.ppat.0030030
- C. Lavazec, C.K. Moreira, G.R. Mair, A.P. Waters, C.J. Janse, and T.J. Templeton, "Analysis of mutant Plasmodium berghei parasites lacking expression of multiple PbCCp genes", Molecular and Biochemical Parasitology, vol. 163, pp. 1-7, 2009. http://dx.doi.org/10.1016/j.molbiopara.2008.09.002
- D. Klug, and F. Frischknecht, "Motility precedes egress of malaria parasites from oocysts", eLife, vol. 6, 2017. http://dx.doi.org/10.7554/eLife.19157
- M. Welch, and M. Way, "Arp2/3-Mediated Actin-Based Motility: A Tail of Pathogen Abuse", Cell Host & Microbe, vol. 14, pp. 242-255, 2013. http://dx.doi.org/10.1016/j.chom.2013.08.011
- J.M. Stevens, E.E. Galyov, and M.P. Stevens, "Actin-dependent movement of bacterial pathogens", Nature Reviews Microbiology, vol. 4, pp. 91-101, 2006. http://dx.doi.org/10.1038/nrmicro1320
- S.M. Rafelski, and J.A. Theriot, "Mechanism of polarization of Listeria monocytogenes surface protein ActA", Molecular Microbiology, vol. 59, pp. 1262-1279, 2006. http://dx.doi.org/10.1111/j.1365-2958.2006.05025.x
- R. Boujemaa-Paterski, E. Gouin, G. Hansen, S. Samarin, C. Le Clainche, D. Didry, P. Dehoux, P. Cossart, C. Kocks, M. Carlier, and D. Pantaloni, "Listeria Protein ActA Mimics WASP Family Proteins: It Activates Filament Barbed End Branching by Arp2/3 Complex", Biochemistry, vol. 40, pp. 11390-11404, 2001. http://dx.doi.org/10.1021/bi010486b
- R. Fattouh, H. Kwon, M.A. Czuczman, J.W. Copeland, L. Pelletier, M.E. Quinlan, A.M. Muise, D.E. Higgins, and J.H. Brumell, "The Diaphanous-Related Formins Promote Protrusion Formation and Cell-to-Cell Spread ofListeria monocytogenes", Journal of Infectious Diseases, vol. 211, pp. 1185-1195, 2014. http://dx.doi.org/10.1093/infdis/jiu546
- T. Rajabian, B. Gavicherla, M. Heisig, S. Müller-Altrock, W. Goebel, S.D. Gray-Owen, and K. Ireton, "The bacterial virulence factor InlC perturbs apical cell junctions and promotes cell-to-cell spread of Listeria", Nature Cell Biology, vol. 11, pp. 1212-1218, 2009. http://dx.doi.org/10.1038/ncb1964
- S. Pust, H. Morrison, J. Wehland, A.S. Sechi, and P. Herrlich, "Listeria monocytogenes exploits ERM protein functions to efficiently spread from cell to cell", The EMBO Journal, vol. 24, pp. 1287-1300, 2005. http://dx.doi.org/10.1038/sj.emboj.7600595
- J.R. Robbins, A.I. Barth, H. Marquis, E.L. de Hostos, W.J. Nelson, and J.A. Theriot, "Listeria monocytogenes Exploits Normal Host Cell Processes to Spread from Cell to Cell✪", The Journal of Cell Biology, vol. 146, pp. 1333-1350, 1999. http://dx.doi.org/10.1083/jcb.146.6.1333
- R. Chong, R. Squires, R. Swiss, and H. Agaisse, "RNAi Screen Reveals Host Cell Kinases Specifically Involved in Listeria monocytogenes Spread from Cell to Cell", PLoS ONE, vol. 6, pp. e23399, 2011. http://dx.doi.org/10.1371/journal.pone.0023399
- J.A. Vazquez-Boland, C. Kocks, S. Dramsi, H. Ohayon, C. Geoffroy, J. Mengaud, and P. Cossart, "Nucleotide sequence of the lecithinase operon of Listeria monocytogenes and possible role of lecithinase in cell-to-cell spread.", Infection and immunity, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1309513
- M.M. Gedde, D.E. Higgins, L.G. Tilney, and D.A. Portnoy, "Role of Listeriolysin O in Cell-to-Cell Spread of Listeria monocytogenes", Infection and Immunity, vol. 68, pp. 999-1003, 2000. http://dx.doi.org/10.1128/iai.68.2.999-1003.2000
- A. Gründling, M.D. Gonzalez, and D.E. Higgins, "Requirement of theListeria monocytogenesBroad-Range Phospholipase PC-PLC during Infection of Human Epithelial Cells", Journal of Bacteriology, vol. 185, pp. 6295-6307, 2003. http://dx.doi.org/10.1128/jb.185.21.6295-6307.2003
- C. Alberti-Segui, K.R. Goeden, and D.E. Higgins, "Differential function of Listeria monocytogenes listeriolysin O and phospholipases C in vacuolar dissolution following cell-to-cell spread", Cellular Microbiology, vol. 9, pp. 179-195, 2007. http://dx.doi.org/10.1111/j.1462-5822.2006.00780.x
- C. Kocks, E. Gouin, M. Tabouret, P. Berche, H. Ohayon, and P. Cossart, "L. monocytogenes-induced actin assembly requires the actA gene product, a surface protein", Cell, vol. 68, pp. 521-531, 1992. http://dx.doi.org/10.1016/0092-8674(92)90188-i
- D.A. Portnoy, V. Auerbuch, and I.J. Glomski, "The cell biology ofListeria monocytogenesinfection", The Journal of Cell Biology, vol. 158, pp. 409-414, 2002. http://dx.doi.org/10.1083/jcb.200205009
- M.L. Bernardini, J. Mounier, H. d'Hauteville, M. Coquis-Rondon, and P.J. Sansonetti, "Identification of icsA, a plasmid locus of Shigella flexneri that governs bacterial intra- and intercellular spread through interaction with F-actin.", Proceedings of the National Academy of Sciences, vol. 86, pp. 3867-3871, 1989. http://dx.doi.org/10.1073/pnas.86.10.3867
- P.J. Sansonetti, J. Mounier, M.C. Prévost, and R. Mège, "Cadherin expression is required for the spread of Shigella flexneri between epithelial cells", Cell, vol. 76, pp. 829-839, 1994. http://dx.doi.org/10.1016/0092-8674(94)90358-1
- Y. Leung, S. Ally, and M.B. Goldberg, "Bacterial Actin Assembly Requires Toca-1 to Relieve N-WASP Autoinhibition", Cell Host & Microbe, vol. 3, pp. 39-47, 2008. http://dx.doi.org/10.1016/j.chom.2007.10.011
- E.A. Bishai, G.S. Sidhu, W. Li, J. Dhillon, A.B. Bohil, R.E. Cheney, J.H. Hartwig, and F.S. Southwick, "Myosin-X facilitatesShigella-induced membrane protrusions and cell-to-cell spread", Cellular Microbiology, vol. 15, pp. 353-367, 2012. http://dx.doi.org/10.1111/cmi.12051
- J.E. Heindl, I. Saran, C. Yi, C.F. Lesser, and M.B. Goldberg, "Requirement for Formin-Induced Actin Polymerization during Spread of Shigella flexneri", Infection and Immunity, vol. 78, pp. 193-203, 2010. http://dx.doi.org/10.1128/IAI.00252-09
- S. Yoshida, Y. Handa, T. Suzuki, M. Ogawa, M. Suzuki, A. Tamai, A. Abe, E. Katayama, and C. Sasakawa, "Microtubule-Severing Activity of Shigella Is Pivotal for Intercellular Spreading", Science, vol. 314, pp. 985-989, 2006. http://dx.doi.org/10.1126/science.1133174
- K.L. Germane, R. Ohi, M.B. Goldberg, and B.W. Spiller, "Structural and Functional Studies Indicate That Shigella VirA Is Not a Protease and Does Not Directly Destabilize Microtubules", Biochemistry, vol. 47, pp. 10241-10243, 2008. http://dx.doi.org/10.1021/bi801533k
- D.M. Monack, and J.A. Theriot, "Actin-based motility is sufficient for bacterial membrane protrusion formation and host cell uptake", Cellular Microbiology, vol. 3, pp. 633-647, 2001. http://dx.doi.org/10.1046/j.1462-5822.2001.00143.x
- M. Fukumatsu, M. Ogawa, S. Arakawa, M. Suzuki, K. Nakayama, S. Shimizu, M. Kim, H. Mimuro, and C. Sasakawa, "Shigella Targets Epithelial Tricellular Junctions and Uses a Noncanonical Clathrin-Dependent Endocytic Pathway to Spread Between Cells", Cell Host & Microbe, vol. 11, pp. 325-336, 2012. http://dx.doi.org/10.1016/j.chom.2012.03.001
- S. Reed, R. Lamason, V. Risca, E. Abernathy, and M. Welch, "Rickettsia Actin-Based Motility Occurs in Distinct Phases Mediated by Different Actin Nucleators", Current Biology, vol. 24, pp. 98-103, 2014. http://dx.doi.org/10.1016/j.cub.2013.11.025
- C.M. Haglund, J.E. Choe, C.T. Skau, D.R. Kovar, and M.D. Welch, "Rickettsia Sca2 is a bacterial formin-like mediator of actin-based motility", Nature Cell Biology, vol. 12, pp. 1057-1063, 2010. http://dx.doi.org/10.1038/ncb2109
- B. Kleba, T.R. Clark, E.I. Lutter, D.W. Ellison, and T. Hackstadt, "Disruption of theRickettsia rickettsiiSca2 Autotransporter Inhibits Actin-Based Motility", Infection and Immunity, vol. 78, pp. 2240-2247, 2010. http://dx.doi.org/10.1128/IAI.00100-10
- R.L. Lamason, E. Bastounis, N.M. Kafai, R. Serrano, J.C. del Álamo, J.A. Theriot, and M.D. Welch, "Rickettsia Sca4 Reduces Vinculin-Mediated Intercellular Tension to Promote Spread", Cell, vol. 167, pp. 670-683.e10, 2016. http://dx.doi.org/10.1016/j.cell.2016.09.023
- S. Thomas, V.L. Popov, and D.H. Walker, "Exit Mechanisms of the Intracellular Bacterium Ehrlichia", PLoS ONE, vol. 5, pp. e15775, 2010. http://dx.doi.org/10.1371/journal.pone.0015775
- D. Mohan Kumar, M. Lin, Q. Xiong, M.J. Webber, C. Kural, and Y. Rikihisa, "EtpE Binding to DNase X Induces Ehrlichial Entry via CD147 and hnRNP-K Recruitment, Followed by Mobilization of N-WASP and Actin", mBio, vol. 6, 2015. http://dx.doi.org/10.1128/mBio.01541-15
- W. Kespichayawattana, S. Rattanachetkul, T. Wanun, P. Utaisincharoen, and S. Sirisinha, "Burkholderia pseudomallei Induces Cell Fusion and Actin-Associated Membrane Protrusion: a Possible Mechanism for Cell-to-Cell Spreading", Infection and Immunity, vol. 68, pp. 5377-5384, 2000. http://dx.doi.org/10.1128/iai.68.9.5377-5384.2000
- E. Benanti, C. Nguyen, and M. Welch, "Virulent Burkholderia Species Mimic Host Actin Polymerases to Drive Actin-Based Motility", Cell, vol. 161, pp. 348-360, 2015. http://dx.doi.org/10.1016/j.cell.2015.02.044
- C.T. French, I.J. Toesca, T. Wu, T. Teslaa, S.M. Beaty, W. Wong, M. Liu, I. Schröder, P. Chiou, M.A. Teitell, and J.F. Miller, "Dissection of the Burkholderia intracellular life cycle using a photothermal nanoblade", Proceedings of the National Academy of Sciences, vol. 108, pp. 12095-12100, 2011. http://dx.doi.org/10.1073/pnas.1107183108
- L.M. Stamm, M.A. Pak, J.H. Morisaki, S.B. Snapper, K. Rottner, S. Lommel, and E.J. Brown, "Role of the WASP family proteins for Mycobacterium marinum actin tail formation", Proceedings of the National Academy of Sciences, vol. 102, pp. 14837-14842, 2005. http://dx.doi.org/10.1073/pnas.0504663102
- L. Volceanov, K. Herbst, M. Biniossek, O. Schilling, D. Haller, T. Nölke, P. Subbarayal, T. Rudel, B. Zieger, and G. Häcker, "Septins Arrange F-Actin-Containing Fibers on the Chlamydia trachomatis Inclusion and Are Required for Normal Release of the Inclusion by Extrusion", mBio, vol. 5, 2014. http://dx.doi.org/10.1128/mBio.01802-14
- E. Lutter, A. Barger, V. Nair, and T. Hackstadt, "Chlamydia trachomatis Inclusion Membrane Protein CT228 Recruits Elements of the Myosin Phosphatase Pathway to Regulate Release Mechanisms", Cell Reports, vol. 3, pp. 1921-1931, 2013. http://dx.doi.org/10.1016/j.celrep.2013.04.027
- M. Kim, M. Kim, and J. Kang, "Involvement of lipid rafts in the budding-like exit of Orientia tsutsugamushi", Microbial Pathogenesis, vol. 63, pp. 37-43, 2013. http://dx.doi.org/10.1016/j.micpath.2013.06.002
- J. Salje, "Orientia tsutsugamushi: A neglected but fascinating obligate intracellular bacterial pathogen", PLOS Pathogens, vol. 13, pp. e1006657, 2017. http://dx.doi.org/10.1371/journal.ppat.1006657
- A. Sturm, R. Amino, C. van de Sand, T. Regen, S. Retzlaff, A. Rennenberg, A. Krueger, J. Pollok, R. Menard, and V.T. Heussler, "Manipulation of Host Hepatocytes by the Malaria Parasite for Delivery into Liver Sinusoids", Science, vol. 313, pp. 1287-1290, 2006. http://dx.doi.org/10.1126/science.1129720
- P. Burda, R. Caldelari, and V.T. Heussler, "Manipulation of the Host Cell Membrane during Plasmodium Liver Stage Egress", mBio, vol. 8, 2017. http://dx.doi.org/10.1128/mBio.00139-17
- S. Graewe, K.E. Rankin, C. Lehmann, C. Deschermeier, L. Hecht, U. Froehlke, R.R. Stanway, and V. Heussler, "Hostile Takeover by Plasmodium: Reorganization of Parasite and Host Cell Membranes during Liver Stage Egress", PLoS Pathogens, vol. 7, pp. e1002224, 2011. http://dx.doi.org/10.1371/journal.ppat.1002224
- K. Baer, C. Klotz, S.H.I. Kappe, T. Schnieder, and U. Frevert, "Release of Hepatic Plasmodium yoelii Merozoites into the Pulmonary Microvasculature", PLoS Pathogens, vol. 3, pp. e171, 2007. http://dx.doi.org/10.1371/journal.ppat.0030171
- T.C. Sudhof, and J. Rizo, "Synaptic Vesicle Exocytosis", Cold Spring Harbor Perspectives in Biology, vol. 3, pp. a005637-a005637, 2011. http://dx.doi.org/10.1101/cshperspect.a005637
- K. Ireton, H. Van Ngo, and M. Bhalla, "Interaction of microbial pathogens with host exocytic pathways", Cellular Microbiology, vol. 20, 2018. http://dx.doi.org/10.1111/cmi.12861
- S.A. Johnston, and R.C. May, "Cryptococcusinteractions with macrophages: evasion and manipulation of the phagosome by a fungal pathogen", Cellular Microbiology, vol. 15, pp. 403-411, 2012. http://dx.doi.org/10.1111/cmi.12067
- J. Chen, K.S. de Felipe, M. Clarke, H. Lu, O.R. Anderson, G. Segal, and H.A. Shuman, "Legionella Effectors That Promote Nonlytic Release from Protozoa", Science, vol. 303, pp. 1358-1361, 2004. http://dx.doi.org/10.1126/science.1094226
- K.S. de Felipe, R.T. Glover, X. Charpentier, O.R. Anderson, M. Reyes, C.D. Pericone, and H.A. Shuman, "Legionella Eukaryotic-Like Type IV Substrates Interfere with Organelle Trafficking", PLoS Pathogens, vol. 4, pp. e1000117, 2008. http://dx.doi.org/10.1371/journal.ppat.1000117
- L. Gomez-Valero, C. Rusniok, C. Cazalet, and C. Buchrieser, "Comparative and Functional Genomics of Legionella Identified Eukaryotic Like Proteins as Key Players in Host?Pathogen Interactions", Frontiers in Microbiology, vol. 2, 2011. http://dx.doi.org/10.3389/fmicb.2011.00208
- X. Shi, P. Halder, H. Yavuz, R. Jahn, and H.A. Shuman, "Direct targeting of membrane fusion by SNARE mimicry: Convergent evolution ofLegionellaeffectors", Proceedings of the National Academy of Sciences, vol. 113, pp. 8807-8812, 2016. http://dx.doi.org/10.1073/pnas.1608755113
- S.G. Berk, R.S. Ting, G.W. Turner, and R.J. Ashburn, "Production of respirable vesicles containing live Legionella pneumophila cells by two Acanthamoeba spp.", Applied and environmental microbiology, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9435080
- S.J. Nolan, M.S. Fu, I. Coppens, and A. Casadevall, "Lipids Affect the Cryptococcus neoformans-Macrophage Interaction and Promote Nonlytic Exocytosis", Infection and Immunity, vol. 85, 2017. http://dx.doi.org/10.1128/IAI.00564-17
- F.H. Santiago-Tirado, M.D. Onken, J.A. Cooper, R.S. Klein, and T.L. Doering, "Trojan Horse Transit Contributes to Blood-Brain Barrier Crossing of a Eukaryotic Pathogen", mBio, vol. 8, 2017. http://dx.doi.org/10.1128/mBio.02183-16
- M. Alvarez, and A. Casadevall, "Phagosome Extrusion and Host-Cell Survival after Cryptococcus neoformans Phagocytosis by Macrophages", Current Biology, vol. 16, pp. 2161-2165, 2006. http://dx.doi.org/10.1016/j.cub.2006.09.061
- H. Ma, J.E. Croudace, D.A. Lammas, and R.C. May, "Expulsion of Live Pathogenic Yeast by Macrophages", Current Biology, vol. 16, pp. 2156-2160, 2006. http://dx.doi.org/10.1016/j.cub.2006.09.032
- S.A. Johnston, and R.C. May, "The Human Fungal Pathogen Cryptococcus neoformans Escapes Macrophages by a Phagosome Emptying Mechanism That Is Inhibited by Arp2/3 Complex-Mediated Actin Polymerisation", PLoS Pathogens, vol. 6, pp. e1001041, 2010. http://dx.doi.org/10.1371/journal.ppat.1001041
- M. Chayakulkeeree, S.A. Johnston, J.B. Oei, S. Lev, P.R. Williamson, C.F. Wilson, X. Zuo, A.L. Leal, M.H. Vainstein, W. Meyer, T.C. Sorrell, R.C. May, and J.T. Djordjevic, "SEC14 is a specific requirement for secretion of phospholipase B1 and pathogenicity of Cryptococcus neoformans", Molecular Microbiology, vol. 80, pp. 1088-1101, 2011. http://dx.doi.org/10.1111/j.1365-2958.2011.07632.x
- A.M. Nicola, E.J. Robertson, P. Albuquerque, L.D.S. Derengowski, and A. Casadevall, "Nonlytic Exocytosis of Cryptococcus neoformans from Macrophages OccursIn Vivoand Is Influenced by Phagosomal pH", mBio, vol. 2, 2011. http://dx.doi.org/10.1128/mBio.00167-11
- A.S. Gilbert, P.I. Seoane, P. Sephton-Clark, A. Bojarczuk, R. Hotham, E. Giurisato, A.R. Sarhan, A. Hillen, G.V. Velde, N.S. Gray, D.R. Alessi, D.L. Cunningham, C. Tournier, S.A. Johnston, and R.C. May, "Vomocytosis of live pathogens from macrophages is regulated by the atypical MAP kinase ERK5", Science Advances, vol. 3, 2017. http://dx.doi.org/10.1126/sciadv.1700898
- M. Hagedorn, K.H. Rohde, D.G. Russell, and T. Soldati, "Infection by Tubercular Mycobacteria Is Spread by Nonlytic Ejection from Their Amoeba Hosts", Science, vol. 323, pp. 1729-1733, 2009. http://dx.doi.org/10.1126/science.1169381
- J. Smith, J. Manoranjan, M. Pan, A. Bohsali, J. Xu, J. Liu, K.L. McDonald, A. Szyk, N. LaRonde-LeBlanc, and L. Gao, "Evidence for Pore Formation in Host Cell Membranes by ESX-1-Secreted ESAT-6 and Its Role in Mycobacterium marinum Escape from the Vacuole", Infection and Immunity, vol. 76, pp. 5478-5487, 2008. http://dx.doi.org/10.1128/IAI.00614-08
- L. Gerstenmaier, R. Pilla, L. Herrmann, H. Herrmann, M. Prado, G.J. Villafano, M. Kolonko, R. Reimer, T. Soldati, J.S. King, and M. Hagedorn, "The autophagic machinery ensures nonlytic transmission of mycobacteria", Proceedings of the National Academy of Sciences, vol. 112, 2015. http://dx.doi.org/10.1073/pnas.1423318112
- E.M. Bijker, S. Borrmann, S.H. Kappe, B. Mordmüller, B.K. Sack, and S.M. Khan, "Novel approaches to whole sporozoite vaccination against malaria", Vaccine, vol. 33, pp. 7462-7468, 2015. http://dx.doi.org/10.1016/j.vaccine.2015.09.095
- O. Kreutzfeld, K. Müller, and K. Matuschewski, "Engineering of Genetically Arrested Parasites (GAPs) For a Precision Malaria Vaccine", Frontiers in Cellular and Infection Microbiology, vol. 7, 2017. http://dx.doi.org/10.3389/fcimb.2017.00198
- M. Singer, and F. Frischknecht, "Time for Genome Editing: Next-Generation Attenuated Malaria Parasites", Trends in Parasitology, vol. 33, pp. 202-213, 2017. http://dx.doi.org/10.1016/j.pt.2016.09.012
ACKNOWLEDGMENTS
The authors thank M. Blackman (The Francis Crick Institute London) and Ö. Günay-Esiyok (Hum-boldt-University Berlin) for helpful discussions. The authors acknowledge funding by the priority programme SPP1580 (AF, GH, MWH, GP) and SFB 1129 (FF) of the Deutsche For-schungsgemeinschaft (DFG). GP is recipient of a DFG Heisen-berg professorship.
COPYRIGHT
© 2018

Pathways of host cell exit by intracellular pathogens by Flieger et al. is licensed under a Creative Commons Attribution 4.0 International License.








