
Reviews:
Microbial Cell, Vol. 3, No. 4, pp. 135 - 146; doi: 10.15698/mic2016.04.489
Signaling pathways and posttranslational modifications of tau in Alzheimer’s disease: the humanization of yeast cells

1 Universität Osnabrück, Fachbereich Biologie/Chemie, AG Genetik, Barbarastr. 11, D-49076 Osnabrück, Germany.
2 Universität Osnabrück, Fachbereich Biologie/Chemie, AG Neurobiologie, Barbarastr. 11, D-49076 Osnabrück, Germany.
Keywords: Saccharomyces cerevisiae, Kluyveromyces lactis, gene expression, neurodegeneration, tauopathies, signal transduction.
Abbreviations:
Aβ – Amyloid beta,
AD – Alzheimer’s disease,
NFT – Neurofibrillary tangle,
PHF – Paired helical filament,
ROS – Reactive oxygen species.
Received originally: 24/01/2016 Accepted: 23/02/2016
Published: 27/03/2016
Correspondence:
Jürgen J. Heinisch, Universität Osnabrück, Fachbereich Biologie/Chemie, AG Neurobiologie, Barbarastr. 11, D-49076 Osnabrück, Germany heinisch@biologie.uni-osnabrueck.de
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Jürgen J. Heinisch and Roland Brandt (2016). Signaling pathways and posttranslational modifications of tau in Alzheimer's disease: the humanization of yeast cells. Microbial Cell 3(4): 135-146.
Abstract
In the past decade, yeast have been frequently employed to study the molecular mechanisms of human neurodegenerative diseases, generally by means of heterologous expression of genes encoding the relevant hallmark proteins. However, it has become evident that substantial posttranslational modifications of many of these proteins are required for the development and progression of potentially disease relevant changes. This is exemplified by the neuronal tau proteins, which are critically involved in a class of neurodegenerative diseases collectively called tauopathies and which includes Alzheimer’s disease (AD) as its most common representative. In the course of the disease, tau changes its phosphorylation state and becomes hyperphosphorylated, gets truncated by proteolytic cleavage, is subject to O-glycosylation, sumoylation, ubiquitinylation, acetylation and some other modifications. This poses the important question, which of these posttranslational modifications are naturally occurring in the yeast model or can be reconstituted by heterologous gene expression. Here, we present an overview on common modifications as they occur in tau during AD, summarize their potential relevance with respect to disease mechanisms and refer to the native yeast enzyme orthologs capable to perform these modifications. We will also discuss potential approaches to humanize yeast in order to create modification patterns resembling the situation in mammalian cells, which could enhance the value of Saccharomyces cerevisiae and Kluyveromyces lactis as disease models.
INTRODUCTION
With the demographic trend of increasing elderly populations in industrialized countries the incidence of different forms of neurodegenerative diseases is presenting a major challenge both personally within the affected families as well as to the social health systems. For example, Alzheimer’s disease (AD) has been estimated to occur in approximately a quarter of people older than 80 years [1]. Albeit the genetic basis of mutations leading to familial forms of AD and other tauopathies have been elucidated, the molecular mechanisms leading to the more common late onset cases of disease are still largely elusive, underlining the necessity for further investigations in different model organisms. In this respect, although obviously lacking a brain with a multicellular neuronal network, simple unicellular yeast are of great value to investigate the effects of overexpression and single point-mutations in genes encoding key proteins on cell physiology and on the function of conserved signaling cascades. Moreover, their increasing application as host organisms for synthetic biology demonstrates the ease by which heterologous genes, such as the ones encoding enzmyes involved in posttranslational modifications, can be expressed and investigated in yeast.
–
Due to its life cycle and its amenability to both classical and advanced molecular genetic techniques, the yeast Saccharomyces cerevisiae has been established as a prevalent eukaryotic model organism [2]. Several excellent reviews have dealt with specific aspects using this model, especially in the study of key AD proteins such as tau and amyloid beta (Aß; see [3][4] and references therein). Here, we will focus on recent advances in the understanding of tauopathies and how baker’s yeast has helped to elucidate disease-relevant mechanisms. We will also refer to mutants and expression systems which could be employed in future studies, demonstrating that the experimental potential has only scarcely been exploited so far. Some advantages of the non-conventional yeast Kluyveromyces lactis, which has not yet been employed to study mechanisms of neurodegenerative diseases, will be briefly addressed with regard to gene redundancy and mitochondrial functions.
TAU HYPERPHOSPHORYLATION AND ITS RELATION TO AD
Histological hallmarks of AD are the accumulation of two different protein aggregates, namely that of the extracellular amyloid precursor protein (APP) peptide Aß within amyloid plaques, and intracellular neurofibrillary tangles (NFTs), which consist of hyperphosphorylated tau proteins that are mainly modified at serine and threonine residues (Fig. 1). It should be noted that NFTs display a better correlation with the cognitive defects associated with AD than amyloid plaques [5][6][7]. Phosphorylated tau proteins in precursors of NFTs either adapt the form of paired helical filaments (PHFs) or straight helical filaments (SFs). The spatiotemporal distribution and aggregation of NFTs within the brain of AD patients and the potential use of tau as a direct target for drug treatment in AD patients have been reviewed [8].
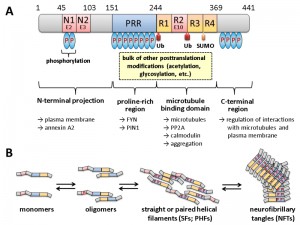 | FIGURE 1: Domain structure and oligomerization of tau. (A) The longest isoform of human tau present in neurons of the central nervous system (CNS) is shown, which differs from other CNS isoforms in the presence or absence of two N-terminal domains (encoded by exons E2 and E3) and one of the repeats in the microtubule interaction domain (R2 encoded by exon E10). For variations in numbering and nomenclature consult [8]. Modifications by ubiquitination (Ub) and SUMOylation (SUMO) are indicated below. Not all phosphorylation sites (P in blue ovals) are shown, but the relative degrees are indicated by the number of ovals. Most of the other covalent modifications take place within the proline-rich region (PRR) and the R1-R4 motif. Some of the cellular structures and proteins for which interaction of the respective domains has been suggested are designated by the arrows shown below. This scheme summarizes informations from several excellent reviews, from which more detailed descriptions of the different modifications and their effect on tau in addition to those mentioned in the text can be obtained: [8][9][10][11][12]. (B) Different oligomerization states of tau are shown. It is believed that the monomeric form fulfills the physiological functions, while soluble oligomers and/or PHFs are responsible for the neurotoxic effects. As indicated, the formation of oligomers is accompanied by increasing degrees of phosphorylation at different sites, as well as by proteolytic trimming (adapted and simplified from [13]). |
–
Originally tau has been identified as a microtubule-associated protein (MAP) in vertebrates, which is abundant in neuronal axons [9][10]. The human tau proteins are encoded by a single MAPT gene on chromosome 17q21. Alternative splicing generates six tau isoforms in the central nervous system, which differ in the presence of three of the 16 encoded exons (Fig. 1). Isoform variation is reflected in different lengths of the N-terminal projection region and the number of microtubule binding repeats in the C-terminal half of the isoforms. Tau belongs to the class of intrinsically disordered proteins, which interact with a large number of partners thereby serving as hubs in cellular protein-protein interaction networks [11]. Modification of tau can render the protein toxic for neurons [12][13][14][15]. In this context, tau neurotoxicity is correlated with several posttranslational modifications, the most prominent being an increased phosphorylation (hyperphosphorylation) at various residues, which affect the strength and spectrum of its interaction with itself and many other cellular components. Based on its amino acid composition, almost 20% of tau protein has the potential to be phosphorylated and tau may thus be considered as a “universal phosphate acceptor” [16][17]. Although it is known that abnormal changes in the phosphorylation state of tau play a key role in the pathogenesis of Alzheimer’s disease, the signaling cascades and specific protein kinases and phosphatases that mediate these alterations are not yet completely understood. It also remains to be elucidated, which are the specific functional changes in tau that are responsible for the pathological consequences. Specific kinases involved in tau phosphorylation which have been studied in the yeast model system have been extensively reviewed [4][18][19] and will be briefly discussed in the respective chapter below.
–
The dominant interaction partners of tau are microtubules and more than 80% of tau is bound to microtubules at physiological conditions [20]. In neurons tau shows a highly dynamic interaction with microtubules [21]. The observed “kiss-and-hop” mechanism explains why tau can have multiple additional interaction partners in different cellular compartments. Thus, tau modifications, e.g. by protein kinases and protein phosphatases, are likely to affect its interactome and thereby the physiological or pathological output (Fig. 2). Recent experimental evidence indicates that soluble oligomeric tau isoforms represent the toxic species [22][23][24]. In contrast, the deposition of higher-order tau aggregates within NFTs may be a cellular response aimed at reducing this toxicity [23][25]. This is reminiscent of the situation in amyloid plaques, where the Aß fragments seem to exert their toxic effects more efficiently in the form of soluble oligomeric precursors, rather than in the aggregates found in the amyloid plaques (reviewed in [3][26]).
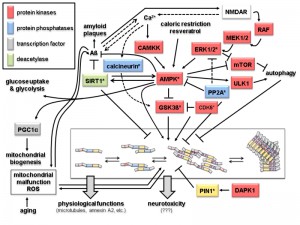 | FIGURE 2: Signaling and modification pathways influencing the intracellular concentration of the toxic oligomeric forms of tau. Arrows indicate activation of enzymes or promotion of a conformational change, lines with bars indicate their inhibition/inactivation. Different protein functions are colour-coded as shown in the upper left hand corner. Schematic representation of tau oligomerization states (central lower part) is as in Fig. 1B. Enzymes with homologs in yeast discussed in the text are marked by an asterisk. Question marks with the neurotoxicity output indicate that the exact molecular mechanisms leading to that condition are not known. Despite the number of different protein functions and signaling cascades displayed in this scheme, not all possible components and interacting networks influencing tau oligomerization are shown. Neither are all possible crosstalks between the depicted components shown (e.g. the protein phosphatase PP2A will also inactivate the upstream components of the ERK1/2 MAPK cascade, calcineurin also acts directly on phosphorylated tau, and PIN1 regulates the activities of GSK3ß and AMPK). In the following, the different signaling pathways which affect tau aggregation are briefly summarized. 1. Activation of tau-phosphorylating protein kinases. The glycogen synthase kinase GSK3ß phosphorylates tau and promotes its aggregation [31]. The kinase activity of GSK3ß is inhibited upon its phosphorylation by the cyclin-dependent protein kinase CDK5, which can also phosphorylate tau, thus either preventing or promoting tau aggregation, respectively [32]. The trimeric AMP-activated kinase complex AMPK phosphorylates and thus inhibits GSK3ß, but can also itself phosphorylate tau and promote its aggregation [33][34]. The catalytic subunit of AMPK itself is activated by phosphorylation, which can be catalyzed either by the calmodulin-dependent protein kinase kinase CAMKK or a redundant pair of mitogen activated protein kinases (MAPKs), designated as extracellular signal recognition kinases ERK1 or ERK2 [35][36]. Whilst CAMKK is activated by Ca2+-bound calmodulin, activation of ERK1/2 is triggered through a conserved MAPK cascade, consisting of the MAPKKs MEK1 and MEK2 and the MAPKKK RAF. Both branches are dependent on the function of the glutamate receptor NMDAR, which triggers an intracellular increase in calcium concentration. This results in the activation of CAMKK and indirectly of RAF through activation of a Ras-GDP/GTP exchange factor (GEF) not depicted here [37]. AMPK, and thereby its inhibitory effect on GSK3ß and tau aggregation, can also be activited by caloric restriction and antioxidants like resveratrol through their influence on the intracellular energy state, i.e. by changing the AMP/ATP ratio [38]. 2. Protein phosphatases. Only two major protein phosphatases which influence tau aggregation are depicted in this scheme, PP2A and calcineurin. Their roles in AD have been reviewed in [39]. Briefly, calcineurin, also called PP2B, activates the tau-kinase GSK3ß and promotes tau aggregation. It can also directly dephosphorylate specific residues in tau and has several other cellular targets, including the NMDAR receptor, relations that are not included here. PP2A has a large variety of substrates, only a few of which are depicted here. Interestingly, it has been found associated to microtubules and has been implicated in many aspects of AD, also reviewed in [39]. In this context, it can desphosphorylate protein kinases in signaling pathways leading to tau aggregation, such as ERK1/2 and AMPK, but also acts directly on phosphorylated tau. 3. Influence of Aß and mitochondrial functions on tau aggregation. The soluble forms of Aß are currently believed to trigger tau aggregation through signaling pathways which still need to be elucidated. Two possible connections are depicted in this scheme. Thus, Aß accumulation leads to an increased calcium flux through NMDAR and thus triggers both the CAMKK and the ERK1/2 mediated activation of AMPK [40]. While Ca2+ concentration and Aß accumulation are interdependent, the latter is prevented by the action of AMPK [41] and by the deacetylase SIRT1 [42]. SIRT1 also inhibits the formation of tau aggregates [43]. It should be noted that SIRT1 and AMPK activities are interdependent in that one can activate the other [42]. AMPK also activates the transcription factor PGC1α, which triggers mitochondrial biogenesis [44]. A by-product of mitochondrial respiration are reactive oxygen species (ROS), whose concentration increase with age and mitochondrial malfunctions. While this leads to aggregation of both Aß and tau, in turn these two pathological hallmarks of AD have been proposed to cause mitochondrial malfunctions [45][46]. 4. Other protein kinases. Indirectly, several other protein kinases involved in different signaling pathways may influence tau aggregation. Thus, ULK1, which is activated by AMPK, promotes autophagy and thereby the degradation of tau aggregates. In the same physiological process, inhibition of the mTOR kinase complex, also mediated by AMPK, prevents its inhibitory effect on autophagy again promoting disposal of tau aggregates [47] [104]. Finally, the death-activated protein kinase DAPK activates the protein isomerase PIN1, which acts on phosphorylated tau and restores its ability to interact with microtubules [48]. |
OTHER POSTTRANSLATIONAL MODIFICATIONS INFLUENCING TAU TOXICITY
In addition to phosphorylation, tau is also subject to a number of other covalent modifications, such as ubiquitination, sumoylation, acetylation, and glycosylation, all affecting its biological activities [27][28] reviewed in [29]. Moreover, different truncated forms of the protein arising from proteolytic cleavage have been implicated in the pathological phenotypes [30][31][32]. Yet, little is known about how these modifications are provoked in a physiological context, how they affect the biological activity of the tau isoforms, and which downstream effectors are involved in mediating the cellular responses. The different modifications and their effects on tau have been extensively reviewed (see references in legend of Fig. 1) and some aspects where yeast may be a good model system will be discussed below.
–
It seems clear, that ubiquitination and subsequent proteasomal degradation is an important mechanism to dispose of toxic tau intermediates. Thus, synaptic accumulation of hyperphosphorylated tau was observed in cells with a defective ubiquitin/proteasome system [33]. Since ubiquitinated tau is mainly found in the insoluble NFTs which accumulate late in AD rather than in the soluble oligomers, ubiquitination may be a way of detoxification. A specific E3 ubiquitin ligase, Axotropin/MARCH7, has been identified in a yeast two-hybrid screen and shown to impair binding of the modified tau to microtubules [34]. Whilst only four lysine residues of tau were found to be ubiquitinated in paired helical filaments of human tau [35], a recent work detected 15 residues in wild-type mouse tau [16]. The discrepancy may be partly due to the fact that most of the lysine residues reported in the latter work are also subject to other modifications, i.e. they can be acetylated and the two modifications are mutually exclusive. This fits well with the observation, that N-acetylation of tau enhances its neurotoxicity, offering the identified acetyltransferase p300 as a therapeutic target [36].
–
With respect to other modifications, hyperphosphorylation of tau and its ubiquitination seem to occur independently (see [37], and references therein). On the other hand, SUMOylation, i.e. the modification by attachment of an ubiquitin-like protein, seems to be triggered by phosporylated tau [38], and vice versa covalent attachment of SUMO to lysine 340 interferes with both phosphorylation and ubiquitination [39]. Oxidative stress, caused by mitochondrial dysfunction as discussed in the next chapter, will also lead to malfunction of the ubiquitin/proteasome system and thus affect tau toxicity [40]. Although the exact regulatory relationships still need to be elucidated, N-glycosylation seems to enhance tau aggregation, while O-GlcNAcetylation has the opposite effect (reviewed in [29][41]).
–
Not only covalent, but also conformational modifications may change the properties, interactions and aggregation propensity of tau. Thus, PIN1, a peptidyl-prolyl cis-trans isomerase, binds to and isomerizes the phosphorylated Thr231 in the proline-rich region of tau (PRR; Fig. 1), thereby restoring its ability to bind microtubules. A death-associated protein kinase, DAPK1, which is activated in the brain of AD patients, was recently identified as a new regulator, which inactivates PIN1 by phosphorylation at Ser71 and thereby contributes to tau pathology (Fig. 2; [42]).
–
Acetylation of proteins can also affect tau aggregation, probably in an indirect manner. Hsp90 acts as a chaperone for tau in its native form, but is kept inactive by acetylation. Active Hsp90 assists in the accumulation of tau aggregates. A deacetylase, HDAC6, catalyzes the activation of Hsp90 and thus enhances tau filament formation [43].
–
Tau is also subject to proteolytic cleavage by several proteases. This includes truncation by caspase-3, which is activated during apoptosis. On the other hand, cleaved tau can itself be pro-apoptotic and trigger a vicious cycle [44]. Tau is also cleaved by the calcium-activated protease calpain, which generates a 17 kDa neurotoxic tau fragment [45]. Moreover, an important role of proteolytic cleavage during the disease process is indicated by the finding that a large portion of tau in PHFs is truncated (Fig. 1; [46]).
–
All these modifications may affect the interaction with more than 70 predicted tau binding partners [35], including the tyrosine kinase fyn, the membrane-associated protein annexin A2, and its effector protein phosphatase PP2A [47][48][49]. Despite this plethora of regulatory systems and interactions, knockout mice lacking a functional tau gene do not display severe defects in brain function or development [50], which suggests the presence of redundant and compensatory mechanims in vertebrates. Analyzing tau interactions and modifications in yeast may therefore provide a powerful tool to obtain novel information on the influence of the interactome in a less redundant environment into which single components may be introduced at will.
SIGNALING PATHWAYS TRIGGERING TAU TOXICITY
In addition to cell death AD is characterized by a change in synaptic connectivity and morphological simplification of dendrites, features jointly referred to as the “neurodegenerative triad” [51]. According to the now well-established amyloid cascade hypothesis Aß acts upstream and mediates the neurodegenerative events, at least partially, through post-translational changes of tau via transmembrane signaling. This may involve both calcineurin signaling mediated by the glutamate receptor NMDAR and signaling of the energy state through SIRT1/AMPK [52][53]. Many components of these signaling cascades are conserved from yeast to humans. For example, signaling through the conserved calmodulin-dependent serine/threonine phosphatase calcineurin [54] triggers activation of the protein kinase GSK3ß, leading to increased tau phosphorylation. A comprehensive scheme of the interconnected signaling pathways affecting tau phosphorylation and mitochondrial functions in neurons is presented in Fig. 2. It should be noted, that these pathways are also connected to mTOR signaling regulating autophagy in the clearance of Aß and tau aggregates, as well as that of dysfunctional mitochondria, whose relation to tauopathies are discussed in the following chapter.
AGING, ENERGY, MITOCHONDRIA AND OXIDATIVE STRESS
Tau phosphorylation and aggregation is both triggered by and triggering itself mitochondrial dysfunctions [55]. Mitochondria serve two important physiological functions relevant to AD development: i) they provide the energy supply especially needed in the mitochondria-rich synapses of the brain, and ii) liberation of pro-apoptotic proteins from the mitochondrial intermembrane space into the cytoplasm, such as holo-cytochrome C, causes caspase activation and apoptotic cell death. It has been suggested that Aß peptides and hyperphosphorylated tau synergistically cause an increased mitochondrial production of reactive oxygen species (ROS), which damage mitochondrial membranes and mtDNA [55]. This leads to more dysfunctional mitochondria and thereby to more ROS production and oxidative stress, which in turn may increase tau phosphorylation and formation of Aß aggregates. Under these conditions hyperphosphorylation of tau seems to be independent of GSK3ß kinase activity. This cycle has been proposed as the “mitochondrial cascade hypothesis” [56][57]. Since mitochondrial dysfunction and ROS production are general hallmarks of the aging process, the hypothesis may also explain the increased incidence of AD with age.
–
The relation between oxidative stress and AD offers the possibility of modulating the effects of ROS by the application of antioxidants, for example the red wine component resveratrol. It has been suggested that resveratrol induces expression of neuroprotective signaling pathways through sirtuin 1 and AMPK acting on Aß and tau at least in a mouse model (Fig. 2) [58]. Yeast cells dispose of the homologs of both sirtuins and AMPK, and Sir2 and the AMPK-homologous SNF1 complex were in fact first described by yeast geneticists and only later on discovered in mammalian cells. This raises the possibility to perform high-throughput screens in yeast for drugs which affect tau modifications (Table 1).
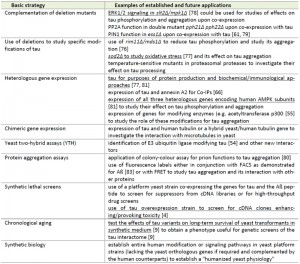 | TABLE 1. Basic approaches to study aspects of tau biology in S. cerevisiae* * aspects also applicable to the alternative non-conventional yeast K. lactis (but not yet examined for tau-related functions) are underlined. Approaches without references are either unpublished results from our laboratory or suggestions for future investigations (see text for details).[4], [9], [54], [55], [61], [66], [76], [77], [78], [79], [80], [81], [82], [83]. |
TAU EXPRESSION IN SACCHAROMYCES CEREVISIAE AND MERITS OF KLUYVEROMYCES LACTIS AS AN ALTERNATIVE YEAST MODEL
Within the past 25 years, the baker’s yeast S. cerevisiae has achieved the status of a perfect unicellular eukaryotic model organism, due to its easy handling both in classical and molecular genetics (reviewed in [2]). There are also triple-shuttle vectors available for fast construction of clones in yeast, amplification in E. coli and expression in neurons [59]. Relevant in the context of this review, heterologous proteins can be cloned and expressed in yeast either constitutively or under the control of the inducible GAL1/10 promoter, allowing the expression of genes whose products are potentially harmful. This has been convincingly demonstrated by the production of functional restriction endonuclease EcoRI in the early days of modern yeast genetics [60].
–
In fact, (over)expression of genes encoding key proteins involved in human dementias proved to be lethal to yeast cells, such as that for synuclein (depending on the genetic background of the yeast strain) and the protein TDP-43 involved in amyotrophic lateral sclerosis (ALS) [61][62]. In contrast, human tau overexpression from the GAL1/10 promoter in yeast, which lacks an endogenous tau homolog, does not produce any detectable phenotype in logarithmically growing wild-type cells [19][47]. Yet, it has been reported that co-expression with the gene encoding synuclein aggravates the toxic effects of overexpression of the latter [63]. More recently, co-expression of tau and α-synuclein genes from genetically stable single-copy yeast integrants confirmed the strong synergistic toxicity [64] which was attributed to the formation of inclusions of α-synuclein and of insoluble cytoplasmic tau aggregates.
–
So far, no yeast deletion mutants which display synthetic lethality with tau overproduction have been reported. However, downregulation of an essential protein, Ess1 (the yeast homolog of the tau modifying human enzyme PIN1; Fig. 2), shows synthetic growth defects with tau overproduction [18]. Recently, a yeast model to study the relation of PIN1/Ess1 and tau has been investigated, showing that the enzyme may inhibit tau phosphorylation [42].
–
In the past decade, expression of tau in S. cerevisiae has been mainly exploited to study the mechanism and effect of hyperphosphorylation. For instance, specific tau mutants expressed in both wild-type and kinase-deficient yeast strains were used to assess their phosphorylation state as well as their in vitro binding capacity to porcine microtubules [65]. These studies revealed that tau in fact gets phosphorylated in yeast by Rim11 (alias Mds1), the ortholog of the human GSK3ß kinase (Fig. 2). Also similar to mammalian tau processing, the activity of this kinase seems to be negatively regulated by the yeast CDK5 ortholog Pho85. Ser409 in the tau protein was specifically identified as a key residue, whose phosphorylation is crucial to produce the aggregated forms in yeast [66]. Thus, although aggregates of hyperphosphorylated tau do form in the yeast system, no growth phenotypes could be detected in logarithmically growing wild-type cultures, indicating that the neurotoxic effects exerted by oligomeric tau in mouse models either depend on a further modification or a specific effector protein, which does not exist in yeast. This offers the exciting possibility of co-expressing human cDNA libraries in a tau overproducing yeast strain in order to screen for such functions, which would render the strains less viable. Moreover, it has been suggested that screens may be directed at chronologically aging yeast cultures [18].
–
As reported above for neurons, Tau aggregation in yeast also occurs under oxidative stress and was shown to be independent of the Rim11/GSK3ß kinase activity [66]. To trigger oxidative stress in this case, S. cerevisiae was either treated with Fe2+, which interferes with mitochondrial function and leads to the production of ROS, or by the use of mutants with defects in mitochondrial proteins such as sod2 or rim1 mutants, which lack a superoxide dismutase or a protein affecting mitochondrial DNA replication, respectively. However, it should be noted that respiratory activity is diminished by glucose-repression in the fermentation-oriented yeast S. cerevisiae, and mitochondria are also less abundant under such conditions. In fact, S. cerevisiae has been selected for thousands of years for its capacity for alcoholic fermentation [67] and with respect to energy production is hardly a physiological equivalent of neurons, which rely on their respiratory metabolism [68].
–
Therefore the milk yeast Kluyveromyces lactis may be a useful alternative since it shares the ease of genetic manipulations with S. cerevisiae, but has a respiratory metabolism more closely resembling that of mammalian cells [69]. For instance, growing under high sugar concentrations (e.g. 2% glucose) mitochondria in K. lactis cells are more readily observed in TEM images as compared to S. cerevisiae ([70] and unpublished results from the laboratory of JJH). K. lactis has also the advantage of not having undergone a whole-genome duplication in evolution and therefore disposes of a much lower redundancy of gene functions, i.e. a single gene deletion will result more readily in a trackable phenotype [69]. Several mutants in homologs of the mammalian signaling pathways depicted in Fig. 2 are already available for K. lactis. Thus, the KlMPK1 gene (homologous to human ERK1/2) has been deleted and shows similar cell wall integrity phenotypes as its S. cerevisiae counterpart SLT2 (alias MPK1; [71]). Mutants within the genes encoding the KlSNF1 complex, the yeast homolog of the AMPK complex, which is composed of the catalytic subunit KlSnf1/Fog2, the ß-subunit KlGal83/Fog1, and the γ-subunit KlSnf4, have been studied for their role in carbohydrate metabolism [72][73]. We dispose of deletion mutants in the genes encoding all three subunits of this complex in K. lactis as well as in the genes encoding the cytoplasmic and the mitochondrial superoxide dismutase, KlSOD1 and KlSOD2 (unpublished results). In principle, this provides the opportunity to study the effects of energy signaling and oxidative stress on tau aggregation in K. lactis, as well as for genetic screens to identify new components involved in this process.
GENETIC SCREENS IN YEAST – THE PAST, THE PRESENT, AND THE FUTURE
For features where respiratory functions are probably irrelevant, S. cerevisiae still represents the yeast system that can be more easily manipulated. It may be of special value when expression of tau-related human genes is required, which lack a yeast homolog or for which functional complementation of the respective deletion by the human counterpart has already been demonstrated. Some basic approaches in which S. cerevisiae may be especially suited to study aspects of tau biology are summarized in Table 1 and some examples will be discussed in the following.
–
Besides the expression/overexpression of different tau isoforms and its lack of phenotypic effects in wild-type yeast discussed above, a number of human genes encoding components of the signaling cascades depicted in Fig. 2 have been successfully expressed, and in many cases shown to functionally complement the respective yeast deletion mutants provided by either of the deletion collections kept in Stanford/USA or the EUROSCARF collection in Frankfurt/Germany. However, it should be noted that caution as to the genetic background of the yeast strain applied for specific purposes may be required, as examplified for the variations in toxic effects of synuclein gene expression [62].
–
One possible application is the expression of the mitogen-activated protein kinases (MAPK) ERK1/ERK2 in yeast. These MAPKs have been proposed to severely affect long-term memory in mouse models [74], and to be activated prior to neuronal tau-related cell death [75]. Although the exact role of ERK1/2 phosphorylation in tau-induced neuronal cell death under physiological conditions is still disputed [76], the yeast system may help to resolve some of the problems. In fact, hyperactive forms of both MAPK isoforms have been expressed in yeast [77], and wild-type forms were shown to be constitutively phosphorylated by a number of yeast MAPKKs [78]. Together with episomal expression of tau, all the tools are thus available to investigate the effects that ERK1/2 variants have on the phosphorylation and aggregation capacity of tau.
–
Furthermore, the phosphorylation state of tau is controlled by the activity of the respective protein kinases (Rim11/Mds1 in yeast and GSK3ß in neurons) and its balance with the activity of protein phosphatases (e.g. PP2A). Mutants in RIM11/MDS1 have already been studied and were shown to decrease the phosphorylation state of tau in yeast [65]. For the catalytic subunit of PP2A two isoforms exist in yeast, encoded by PPH21 and PPH22 [79]. Double pph21 pph22 deletion mutants could thus be used for expression of a codon-optimized gene encoding the human PP2A catalytic subunit, to test its effect on tau phosphorylation.
–
Regarding the conformational changes in phosphorylated tau restoring its capacity to bind to microtubules, the human PIN1 ortholog functionally complements a yeast deletion in the essential gene ESS1 [80]. Its relation to tau biology has been studied in a yeast model [42]. In this context, the yeast model system presents two major obstacles: i) tau does not bind to yeast tubulin and thus has to be extracted from yeast cells and tested separately for its binding capacity to porcine microtubules [65], and ii) to determine the fraction of insoluble tau aggregates, they also have to be laboriously isolated from yeast cells and quantified. With the ease of yeast genetics, both problems may be overcome. The failure of tau to bind to yeast tubulin is most likely due to sequence differences between yeast α-tubulin and its mammalian homolog, located at the proposed tau-binding site [81]. It may thus be possible to produce chimeric yeast/human microtubules in such a strain which can be employed for in vivo binding assays. On the other hand, amyloid-forming capacity has been studied for a variety of prion proteins by using a red/white colony colour assay for aggregation capacity (reviewed in [82]), which may also be applied to tau.
–
As a last example of using established yeast mutants and chimeric yeast/human enzymes the yeast SNF1 complex was discovered years before its human homolog AMPK, both being involved in energy signaling (see [83], and references therein). As shown in Fig. 2, besides being a target of the protein phosphatase PP2A in humans, it mediates the phosphorylation state of tau both directly and indirectly through regulation of GSK3ß kinase activity. Deletion mutants in all three subunits of the complex are readily available in different yeast strains and where shown to affect not only carbohydrate metabolism but also a variety of other signal transduction mechanisms [83]. Importantly, mammalian orthologs of the catalytic α-subunit did not complement the yeast snf1 defects, but co-expression of all three genes encoding the complex did [84]. This humanized yeast may thus be employed to study the relation of AMPK and tau phosphorylation.
–
Of the experimental approaches listed in Table 1, yeast two-hybrid screens have already been applied to identify components of the tau modification network, e.g. the E3 ubiquitin ligase Axotropin/MARCH7 [34]. As a complementary strategy, we have expressed tagged versions of wild-type and a mutant tau (R406W) together with annexin A2 in yeast to confirm their interaction by co-immunoprecipitations, thus evading interferences which might be caused by larger neuron-specific protein complexes [47].
–
In general heterologous gene expression in yeast is a powerful tool. Thus, it has also served as a means to produce enough tau protein to obtain new monoclonal antibodies which can be used to differentiate the oligomerization state [85]. In the future, it may be employed to set up high-throughput screens for drugs affecting enzyme activities, such as that of the above mentioned acetyltransferase p300 [36], or to identify the enzyme that mediates O-glycosylation of cytoplasmic tau. For the latter kind of assays it would be very useful to find more synthetic phenotypes of tau (over)expression with different yeast deletion mutants, to allow for screens with positive selection of plasmids from cDNA libraries or small-molecule drugs affecting tau stability and aggregation.
OUTLOOK
Although tau biology is a highly competitive field and has seen a considerable amount of research, crucial aspects of its upstream regulators and its downstream effectors ultimately leading to neuronal toxicity in the brain of AD patients remain to be elucidated. The yeast S. cerevisiae has been widely exploited as a platform to discover the basis of toxicity of α-synuclein and as a vehicle for high-thoughput therapeutic drug screens [86]. Partly due to the lack of toxicity of tau gene expression in yeast cells, they have not yet been used to a similar extent for studying tauopathies and the underlying modification and signaling networks. In fact, as outlined above, yeast so far mainly served as an expression vehicle and to investigate only some aspects of tau hyperphosphorylation and aggregation. In this review we have pointed out several lines of research which could significantly profit from the yeast model and that many tools to this end are already at hand. These perspectives are even further broadened by the growing use of yeast cells in synthetic biology, so far culminating in the expression of as much as 23 genes from organisms belonging to all biological kingdoms in a modified platform yeast strain, used to establish the entire heterologous pathway for opiate production [87].
–
Thus, given the ease of genetic manipulation, with countless expression vectors for all purposes and the availability of deletion collections for the entire yeast genome, it is safe to say that investigations of tau biology in yeast have only just begun and may even profit from some non-conventional yeast models. Clearly, a close collaboration of “classical” tau researchers with yeast geneticists will be highly beneficial in this endeavor.
References
- . , "2015 Alzheimer's disease facts and figures", Alzheimer's & Dementia, vol. 11, pp. 332-384, 2015. http://dx.doi.org/10.1016/j.jalz.2015.02.003
- J.J. Heinisch, "Baker's yeast as a tool for the development of antifungal kinase inhibitors—targeting protein kinase C and the cell integrity pathway", Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, vol. 1754, pp. 171-182, 2005. http://dx.doi.org/10.1016/j.bbapap.2005.07.032
- B. Moosavi, B. Mousavi, and I.G. Macreadie, "Yeast Model of Amyloid-β and Tau Aggregation in Alzheimer’s Disease", Journal of Alzheimer's Disease, vol. 47, pp. 9-16, 2015. http://dx.doi.org/10.3233/JAD-150173
- M. Verduyckt, H. Vignaud, T. Bynens, J. Van den Brande, V. Franssens, C. Cullin, and J. Winderickx, "Yeast as a Model for Alzheimer’s Disease: Latest Studies and Advanced Strategies", Systems Biology of Alzheimer's Disease, pp. 197-215, 2016. http://dx.doi.org/10.1007/978-1-4939-2627-5_11
- A.L. Guillozet, S. Weintraub, D.C. Mash, and M.M. Mesulam, "Neurofibrillary Tangles, Amyloid, and Memory in Aging and Mild Cognitive Impairment", Archives of Neurology, vol. 60, pp. 729, 2003. http://dx.doi.org/10.1001/archneur.60.5.729
- P.T. Nelson, I. Alafuzoff, E.H. Bigio, C. Bouras, H. Braak, N.J. Cairns, R.J. Castellani, B.J. Crain, P. Davies, K.D. Tredici, C. Duyckaerts, M.P. Frosch, V. Haroutunian, P.R. Hof, C.M. Hulette, B.T. Hyman, T. Iwatsubo, K.A. Jellinger, G.A. Jicha, E. Kövari, W.A. Kukull, J.B. Leverenz, S. Love, I.R. Mackenzie, D.M. Mann, E. Masliah, A.C. McKee, T.J. Montine, J.C. Morris, J.A. Schneider, J.A. Sonnen, D.R. Thal, J.Q. Trojanowski, J.C. Troncoso, T. Wisniewski, R.L. Woltjer, and T.G. Beach, "Correlation of Alzheimer Disease Neuropathologic Changes With Cognitive Status: A Review of the Literature", Journal of Neuropathology & Experimental Neurology, vol. 71, pp. 362-381, 2012. http://dx.doi.org/10.1097/NEN.0b013e31825018f7
- A. Serrano-Pozo, M.P. Frosch, E. Masliah, and B.T. Hyman, "Neuropathological Alterations in Alzheimer Disease", Cold Spring Harbor Perspectives in Medicine, vol. 1, pp. a006189-a006189, 2011. http://dx.doi.org/10.1101/cshperspect.a006189
- L. Bakota, and R. Brandt, "Tau Biology and Tau-Directed Therapies for Alzheimer’s Disease", Drugs, vol. 76, pp. 301-313, 2016. http://dx.doi.org/10.1007/s40265-015-0529-0
- L.I. Binder, A. Frankfurter, and L.I. Rebhun, "The distribution of tau in the mammalian central nervous system.", The Journal of cell biology, 1985. http://www.ncbi.nlm.nih.gov/pubmed/3930508
- L. Dehmelt, and S. Halpain, "Array", Genome Biology, vol. 6, pp. 204, 2004. http://dx.doi.org/10.1186/gb-2004-6-1-204
- V.N. Uversky, "Intrinsically disordered proteins and their (disordered) proteomes in neurodegenerative disorders", Frontiers in Aging Neuroscience, vol. 7, 2015. http://dx.doi.org/10.3389/fnagi.2015.00018
- T. Fath, J. Eidenmüller, and R. Brandt, "Tau-mediated cytotoxicity in a pseudohyperphosphorylation model of Alzheimer's disease.", The Journal of neuroscience : the official journal of the Society for Neuroscience, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12427828
- M. Rapoport, H.N. Dawson, L.I. Binder, M.P. Vitek, and A. Ferreira, "Tau is essential to β-amyloid-induced neurotoxicity", Proceedings of the National Academy of Sciences, vol. 99, pp. 6364-6369, 2002. http://dx.doi.org/10.1073/pnas.092136199
- M. Rapoport, and A. Ferreira, "PD98059 Prevents Neurite Degeneration Induced by Fibrillar β‐Amyloid in Mature Hippocampal Neurons", Journal of Neurochemistry, vol. 74, pp. 125-133, 2000. http://dx.doi.org/10.1046/j.1471-4159.2000.0740125.x
- C. Tackenberg, and R. Brandt, "Divergent Pathways Mediate Spine Alterations and Cell Death Induced by Amyloid-β, Wild-Type Tau, and R406W Tau", The Journal of Neuroscience, vol. 29, pp. 14439-14450, 2009. http://dx.doi.org/10.1523/JNEUROSCI.3590-09.2009
- M. Morris, G.M. Knudsen, S. Maeda, J.C. Trinidad, A. Ioanoviciu, A.L. Burlingame, and L. Mucke, "Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice", Nature Neuroscience, vol. 18, pp. 1183-1189, 2015. http://dx.doi.org/10.1038/nn.4067
- W.H. Stoothoff, and G.V. Johnson, "Tau phosphorylation: physiological and pathological consequences", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1739, pp. 280-297, 2005. http://dx.doi.org/10.1016/j.bbadis.2004.06.017
- A. De Vos, J. Anandhakumar, J. Van den Brande, M. Verduyckt, V. Franssens, J. Winderickx, and E. Swinnen, "Yeast as a Model System to Study Tau Biology", International Journal of Alzheimer’s Disease, vol. 2011, 2011. http://dx.doi.org/10.4061/2011/428970
- J. Winderickx, C. Delay, A. De Vos, H. Klinger, K. Pellens, T. Vanhelmont, F. Van Leuven, and P. Zabrocki, "Protein folding diseases and neurodegeneration: Lessons learned from yeast", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1783, pp. 1381-1395, 2008. http://dx.doi.org/10.1016/j.bbamcr.2008.01.020
- C. Weissmann, H. Reyher, A. Gauthier, H. Steinhoff, W. Junge, and R. Brandt, "Microtubule Binding and Trapping at the Tip of Neurites Regulate Tau Motion in Living Neurons", Traffic, vol. 10, pp. 1655-1668, 2009. http://dx.doi.org/10.1111/j.1600-0854.2009.00977.x
- D. Janning, M. Igaev, F. Sündermann, J. Brühmann, O. Beutel, J.J. Heinisch, L. Bakota, J. Piehler, W. Junge, and R. Brandt, "Single-molecule tracking of tau reveals fast kiss-and-hop interaction with microtubules in living neurons", Molecular Biology of the Cell, vol. 25, pp. 3541-3551, 2014. http://dx.doi.org/10.1091/mbc.E14-06-1099
- J.E. Gerson, U. Sengupta, C.A. Lasagna-Reeves, M.J. Guerrero-Muñoz, J. Troncoso, and R. Kayed, "Characterization of tau oligomeric seeds in progressive supranuclear palsy", Acta Neuropathologica Communications, vol. 2, 2014. http://dx.doi.org/10.1186/2051-5960-2-73
- T.L. Spires-Jones, K.J. Kopeikina, R.M. Koffie, A. de Calignon, and B.T. Hyman, "Are Tangles as Toxic as They Look?", Journal of Molecular Neuroscience, vol. 45, pp. 438-444, 2011. http://dx.doi.org/10.1007/s12031-011-9566-7
- T.L. Spires-Jones, W.H. Stoothoff, A. de Calignon, P.B. Jones, and B.T. Hyman, "Tau pathophysiology in neurodegeneration: a tangled issue", Trends in Neurosciences, vol. 32, pp. 150-159, 2009. http://dx.doi.org/10.1016/j.tins.2008.11.007
- J.E. Gerson, and R. Kayed, "Formation and Propagation of Tau Oligomeric Seeds", Frontiers in Neurology, vol. 4, 2013. http://dx.doi.org/10.3389/fneur.2013.00093
- G.M. Shankar, B.L. Bloodgood, M. Townsend, D.M. Walsh, D.J. Selkoe, and B.L. Sabatini, "Natural Oligomers of the Alzheimer Amyloid-β Protein Induce Reversible Synapse Loss by Modulating an NMDA-Type Glutamate Receptor-Dependent Signaling Pathway", The Journal of Neuroscience, vol. 27, pp. 2866-2875, 2007. http://dx.doi.org/10.1523/JNEUROSCI.4970-06.2007
- L. Martin, X. Latypova, and F. Terro, "Post-translational modifications of tau protein: Implications for Alzheimer's disease", Neurochemistry International, vol. 58, pp. 458-471, 2011. http://dx.doi.org/10.1016/j.neuint.2010.12.023
- Y. Zhu, X. Shan, S.A. Yuzwa, and D.J. Vocadlo, "The Emerging Link between O-GlcNAc and Alzheimer Disease", Journal of Biological Chemistry, vol. 289, pp. 34472-34481, 2014. http://dx.doi.org/10.1074/jbc.R114.601351
- S.N. Fontaine, J.J. Sabbagh, J. Baker, C.R. Martinez-Licha, A. Darling, and C.A. Dickey, "Cellular factors modulating the mechanism of tau protein aggregation", Cellular and Molecular Life Sciences, vol. 72, pp. 1863-1879, 2015. http://dx.doi.org/10.1007/s00018-015-1839-9
- P. Flores-RodrÃguez, M.A. Ontiveros-Torres, M.C. Cárdenas-Aguayo, J.P. Luna-Arias, M.A. Meraz-RÃos, A. Viramontes-Pintos, C.R. Harrington, C.M. Wischik, R. Mena, B. Florán-Garduño, and J. Luna-Muñoz, "The relationship between truncation and phosphorylation at the C-terminus of tau protein in the paired helical filaments of Alzheimer's disease", Frontiers in Neuroscience, vol. 9, 2015. http://dx.doi.org/10.3389/fnins.2015.00033
- D. Kanmert, A. Cantlon, C.R. Muratore, M. Jin, T.T. O'Malley, G. Lee, T.L. Young-Pearse, D.J. Selkoe, and D.M. Walsh, "C-Terminally Truncated Forms of Tau, But Not Full-Length Tau or Its C-Terminal Fragments, Are Released from Neurons Independently of Cell Death", Journal of Neuroscience, vol. 35, pp. 10851-10865, 2015. http://dx.doi.org/10.1523/JNEUROSCI.0387-15.2015
- K. Paholikova, B. Salingova, A. Opattova, R. Skrabana, P. Majerova, N. Zilka, B. Kovacech, M. Zilkova, P. Barath, and M. Novak, "N-terminal truncation of microtubule associated protein tau dysregulates its cellular localization.", Journal of Alzheimer's disease : JAD, 2015. http://www.ncbi.nlm.nih.gov/pubmed/25147106
- H. Tai, A. Serrano-Pozo, T. Hashimoto, M.P. Frosch, T.L. Spires-Jones, and B.T. Hyman, "The Synaptic Accumulation of Hyperphosphorylated Tau Oligomers in Alzheimer Disease Is Associated With Dysfunction of the Ubiquitin-Proteasome System", The American Journal of Pathology, vol. 181, pp. 1426-1435, 2012. http://dx.doi.org/10.1016/j.ajpath.2012.06.033
- K. Flach, E. Ramminger, I. Hilbrich, A. Arsalan-Werner, F. Albrecht, L. Herrmann, M. Goedert, T. Arendt, and M. Holzer, "Axotrophin/MARCH7 acts as an E3 ubiquitin ligase and ubiquitinates tau protein in vitro impairing microtubule binding", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 1527-1538, 2014. http://dx.doi.org/10.1016/j.bbadis.2014.05.029
- Y. Ihara, M. Morishima-Kawashima, and R. Nixon, "The Ubiquitin-Proteasome System and the Autophagic-Lysosomal System in Alzheimer Disease", Cold Spring Harbor Perspectives in Medicine, vol. 2, pp. a006361-a006361, 2012. http://dx.doi.org/10.1101/cshperspect.a006361
- S. Min, X. Chen, T.E. Tracy, Y. Li, Y. Zhou, C. Wang, K. Shirakawa, S.S. Minami, E. Defensor, S.A. Mok, P.D. Sohn, B. Schilling, X. Cong, L. Ellerby, B.W. Gibson, J. Johnson, N. Krogan, M. Shamloo, J. Gestwicki, E. Masliah, E. Verdin, and L. Gan, "Critical role of acetylation in tau-mediated neurodegeneration and cognitive deficits", Nature Medicine, vol. 21, pp. 1154-1162, 2015. http://dx.doi.org/10.1038/nm.3951
- J.M. Deger, J.E. Gerson, and R. Kayed, "The interrelationship of proteasome impairment and oligomeric intermediates in neurodegeneration", Aging Cell, vol. 14, pp. 715-724, 2015. http://dx.doi.org/10.1111/acel.12359
- V. Dorval, and P.E. Fraser, "Small Ubiquitin-like Modifier (SUMO) Modification of Natively Unfolded Proteins Tau and α-Synuclein", Journal of Biological Chemistry, vol. 281, pp. 9919-9924, 2006. http://dx.doi.org/10.1074/jbc.M510127200
- H. Luo, Y. Xia, X. Shu, Z. Liu, Y. Feng, X. Liu, G. Yu, G. Yin, Y. Xiong, K. Zeng, J. Jiang, K. Ye, X. Wang, and J. Wang, "SUMOylation at K340 inhibits tau degradation through deregulating its phosphorylation and ubiquitination", Proceedings of the National Academy of Sciences, vol. 111, pp. 16586-16591, 2014. http://dx.doi.org/10.1073/pnas.1417548111
- B.M. Riederer, G. Leuba, and Z. ElHajj, "Oxidation and ubiquitination in neurodegeneration", Experimental Biology and Medicine, vol. 238, pp. 519-524, 2013. http://dx.doi.org/10.1177/1535370213488484
- S. Schedin‐Weiss, B. Winblad, and L.O. Tjernberg, "The role of protein glycosylation in Alzheimer disease", The FEBS Journal, vol. 281, pp. 46-62, 2013. http://dx.doi.org/10.1111/febs.12590
- A. De Vos, . , T. Bynens, J. Rosseels, C. Coun, J. Ring, F. Madeo, M. Galas, J. Winderickx, and V. Franssens, "The peptidyl prolyl cis/trans isomerase Pin1/Ess1 inhibits phosphorylation and toxicity of tau in a yeast model for Alzheimer’s disease", AIMS Molecular Science, vol. 2, pp. 144-160, 2015. http://dx.doi.org/10.3934/molsci.2015.2.144
- C. Cook, T.F. Gendron, K. Scheffel, Y. Carlomagno, J. Dunmore, M. DeTure, and L. Petrucelli, "Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation", Human Molecular Genetics, vol. 21, pp. 2936-2945, 2012. http://dx.doi.org/10.1093/hmg/dds125
- C. Chung, Y. Song, I. Kim, W. Yoon, B. Ryu, D. Jo, H. Woo, Y. Kwon, H. Kim, B. Gwag, I. Mook-Jung, and Y. Jung, "Proapoptotic Effects of Tau Cleavage Product Generated by Caspase-3", Neurobiology of Disease, vol. 8, pp. 162-172, 2001. http://dx.doi.org/10.1006/nbdi.2000.0335
- S. Park, and A. Ferreira, "The Generation of a 17 kDa Neurotoxic Fragment: An Alternative Mechanism by which Tau Mediates β-Amyloid-Induced Neurodegeneration", The Journal of Neuroscience, vol. 25, pp. 5365-5375, 2005. http://dx.doi.org/10.1523/JNEUROSCI.1125-05.2005
- B. Kovacech, and M. Novak, "Tau Truncation is a Productive Posttranslational Modification of Neurofibrillary Degeneration in Alzheimers Disease", Current Alzheimer Research, vol. 7, pp. 708-716, 2010. http://dx.doi.org/10.2174/156720510793611556
- A. Gauthier-Kemper, C. Weissmann, N. Golovyashkina, Z. Sebö-Lemke, G. Drewes, V. Gerke, J.J. Heinisch, and R. Brandt, "The frontotemporal dementia mutation R406W blocks tau’s interaction with the membrane in an annexin A2–dependent manner", Journal of Cell Biology, vol. 192, pp. 647-661, 2011. http://dx.doi.org/10.1083/jcb.201007161
- G. Lee, S.T. Newman, D.L. Gard, H. Band, and G. Panchamoorthy, "Tau interacts with src-family non-receptor tyrosine kinases.", Journal of cell science, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9763511
- J. Sontag, and E. Sontag, "Protein phosphatase 2A dysfunction in Alzheimer’s disease", Frontiers in Molecular Neuroscience, vol. 7, 2014. http://dx.doi.org/10.3389/fnmol.2014.00016
- A. Harada, K. Oguchi, S. Okabe, J. Kuno, S. Terada, T. Ohshima, R. Sato-Yoshitake, Y. Takei, T. Noda, and N. Hirokawa, "Altered microtubule organization in small-calibre axons of mice lacking tau protein", Nature, vol. 369, pp. 488-491, 1994. http://dx.doi.org/10.1038/369488a0
- H. Wu, E. Hudry, T. Hashimoto, K. Kuchibhotla, A. Rozkalne, Z. Fan, T. Spires-Jones, H. Xie, M. Arbel-Ornath, C.L. Grosskreutz, B.J. Bacskai, and B.T. Hyman, "Amyloid β Induces the Morphological Neurodegenerative Triad of Spine Loss, Dendritic Simplification, and Neuritic Dystrophies through Calcineurin Activation", The Journal of Neuroscience, vol. 30, pp. 2636-2649, 2010. http://dx.doi.org/10.1523/JNEUROSCI.4456-09.2010
- D.L. Smith, J. Pozueta, B. Gong, O. Arancio, and M. Shelanski, "Reversal of long-term dendritic spine alterations in Alzheimer disease models", Proceedings of the National Academy of Sciences, vol. 106, pp. 16877-16882, 2009. http://dx.doi.org/10.1073/pnas.0908706106
- C. Tackenberg, S. Grinschgl, A. Trutzel, A.C. Santuccione, M.C. Frey, U. Konietzko, J. Grimm, R. Brandt, and R.M. Nitsch, "NMDA receptor subunit composition determines beta-amyloid-induced neurodegeneration and synaptic loss", Cell Death & Disease, vol. 4, pp. e608-e608, 2013. http://dx.doi.org/10.1038/cddis.2013.129
- G. Caraveo, P.K. Auluck, L. Whitesell, C.Y. Chung, V. Baru, E.V. Mosharov, X. Yan, M. Ben-Johny, M. Soste, P. Picotti, H. Kim, K.A. Caldwell, G.A. Caldwell, D. Sulzer, D.T. Yue, and S. Lindquist, "Calcineurin determines toxic versus beneficial responses to α-synuclein", Proceedings of the National Academy of Sciences, vol. 111, 2014. http://dx.doi.org/10.1073/pnas.1413201111
- A. Eckert, R. Nisbet, A. Grimm, and J. Götz, "March separate, strike together — Role of phosphorylated TAU in mitochondrial dysfunction in Alzheimer's disease", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1842, pp. 1258-1266, 2014. http://dx.doi.org/10.1016/j.bbadis.2013.08.013
- F.M. Albrekkan, and M. Kelly-Worden, "Mitochondrial Dysfunction and Alzheimer’s Disease", Open Journal of Endocrine and Metabolic Diseases, vol. 03, pp. 14-19, 2013. http://dx.doi.org/10.4236/ojemd.2013.32A003
- K. Friedland-Leuner, C. Stockburger, I. Denzer, G.P. Eckert, and W.E. Müller, "Mitochondrial Dysfunction", Progress in Molecular Biology and Translational Science, pp. 183-210, 2014. http://dx.doi.org/10.1016/B978-0-12-394625-6.00007-6
- D. Porquet, C. Griñán-Ferré, I. Ferrer, A. Camins, C. Sanfeliu, J. Del Valle, and M. Pallàs, "Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer's disease.", Journal of Alzheimer's disease : JAD, 2014. http://www.ncbi.nlm.nih.gov/pubmed/25024312
- L. Bakota, R. Brandt, and J.J. Heinisch, "Triple mammalian/yeast/bacterial shuttle vectors for single and combined Lentivirus- and Sindbis virus-mediated infections of neurons", Molecular Genetics and Genomics, vol. 287, pp. 313-324, 2012. http://dx.doi.org/10.1007/s00438-012-0680-1
- G. Barnes, and J. Rine, "Regulated expression of endonuclease EcoRI in Saccharomyces cerevisiae: nuclear entry and biological consequences.", Proceedings of the National Academy of Sciences of the United States of America, 1985. http://www.ncbi.nlm.nih.gov/pubmed/2983340
- M.E. Jackrel, and J. Shorter, "Potentiated Hsp104 variants suppress toxicity of diverse neurodegenerative disease-linked proteins", Disease Models & Mechanisms, 2014. http://dx.doi.org/10.1242/dmm.016113
- N. Sharma, K.A. Brandis, S.K. Herrera, B.E. Johnson, T. Vaidya, R. Shrestha, and S.K. DebBurman, "α-Synuclein Budding Yeast Model: Toxicity Enhanced by Impaired Proteasome and Oxidative Stress", Journal of Molecular Neuroscience, vol. 28, pp. 161-178, 2006. http://dx.doi.org/10.1385/JMN:28:2:161
- P. Zabrocki, K. Pellens, T. Vanhelmont, T. Vandebroek, G. Griffioen, S. Wera, F. Van Leuven, and J. Winderickx, "Characterization of α‐synuclein aggregation and synergistic toxicity with protein tau in yeast", The FEBS Journal, vol. 272, pp. 1386-1400, 2005. http://dx.doi.org/10.1111/j.1742-4658.2005.04571.x
- G. Ciaccioli, A. Martins, C. Rodrigues, H. Vieira, and P. Calado, "A Powerful Yeast Model to Investigate the Synergistic Interaction of α-Synuclein and Tau in Neurodegeneration", PLoS ONE, vol. 8, pp. e55848, 2013. http://dx.doi.org/10.1371/journal.pone.0055848
- T. Vandebroek, D. Terwel, T. Vanhelmont, M. Gysemans, C. Van Haesendonck, Y. Engelborghs, J. Winderickx, and F. Van Leuven, "Microtubule Binding and Clustering of Human Tau-4R and Tau-P301L Proteins Isolated from Yeast Deficient in Orthologues of Glycogen Synthase Kinase-3β or cdk5", Journal of Biological Chemistry, vol. 281, pp. 25388-25397, 2006. http://dx.doi.org/10.1074/jbc.m602792200
- T. Vanhelmont, T. Vandebroek, A. De Vos, D. Terwel, K. Lemaire, J. Anandhakumar, V. Franssens, E. Swinnen, F. Van Leuven, and J. Winderickx, "Serine-409 phosphorylation and oxidative damage define aggregation of human protein tau in yeast", FEMS Yeast Research, vol. 10, pp. 992-1005, 2010. http://dx.doi.org/10.1111/j.1567-1364.2010.00662.x
- S. Dashko, N. Zhou, C. Compagno, and J. Piškur, "Why, when, and how did yeast evolve alcoholic fermentation?", FEMS Yeast Research, vol. 14, pp. 826-832, 2014. http://dx.doi.org/10.1111/1567-1364.12161
- J.P. Bolaños, A. Almeida, and S. Moncada, "Glycolysis: a bioenergetic or a survival pathway?", Trends in Biochemical Sciences, vol. 35, pp. 145-149, 2010. http://dx.doi.org/10.1016/j.tibs.2009.10.006
- R. Rodicio, and J.J. Heinisch, "Yeast on the milky way: genetics, physiology and biotechnology of Kluyveromyces lactis", Yeast, vol. 30, pp. 165-177, 2013. http://dx.doi.org/10.1002/yea.2954
- K. Backhaus, C.J. Heilmann, A.G. Sorgo, G. Purschke, C.G. de Koster, F.M. Klis, and J.J. Heinisch, "A systematic study of the cell wall composition of Kluyveromyces lactis", Yeast, vol. 27, pp. 647-660, 2010. http://dx.doi.org/10.1002/yea.1781
- L. Kirchrath, A. Lorberg, H. Schmitz, U. Gengenbacher, and J.J. Heinisch, "Comparative Genetic and Physiological Studies of the MAP Kinase Mpk1p from Kluyveromyces lactis and Saccharomyces cerevisiae", Journal of Molecular Biology, vol. 300, pp. 743-758, 2000. http://dx.doi.org/10.1006/jmbi.2000.3916
- P. Goffrini, A. Ficarelli, C. Donnini, T. Lodi, P.P. Puglisi, and I. Ferrero, "FOG1 and FOG2 genes, required for the transcriptional activation of glucose-repressible genes of Kluyveromyces lactis , are homologous to GAL83 and SNF1 of Saccharomyces cerevisiae", Current Genetics, vol. 29, pp. 316-326, 1996. http://dx.doi.org/10.1007/s002940050052
- C. Wiedemuth, and K.D. Breunig, "Role of Snf1p in Regulation of Intracellular Sorting of the Lactose and Galactose Transporter Lac12p in Kluyveromyces lactis", Eukaryotic Cell, vol. 4, pp. 716-721, 2005. http://dx.doi.org/10.1128/EC.4.4.716-721.2005
- S. Peng, Y. Zhang, J. Zhang, H. Wang, and B. Ren, "ERK in Learning and Memory: A Review of Recent Research", International Journal of Molecular Sciences, vol. 11, pp. 222-232, 2010. http://dx.doi.org/10.3390/ijms11010222
- G. Amadoro, M.T. Ciotti, M. Costanzi, V. Cestari, P. Calissano, and N. Canu, "NMDA receptor mediates tau-induced neurotoxicity by calpain and ERK/MAPK activation", Proceedings of the National Academy of Sciences, vol. 103, pp. 2892-2897, 2006. http://dx.doi.org/10.1073/pnas.0511065103
- A. Noël, I. Poitras, J. Julien, F.R. Petry, F. Morin, J. Charron, and E. Planel, "ERK (MAPK) does not phosphorylate tau under physiological conditions in vivo or in vitro", Neurobiology of Aging, vol. 36, pp. 901-902, 2015. http://dx.doi.org/10.1016/j.neurobiolaging.2014.11.005
- V. Levin-Salomon, K. Kogan, N.G. Ahn, O. Livnah, and D. Engelberg, "Isolation of Intrinsically Active (MEK-independent) Variants of the ERK Family of Mitogen-activated Protein (MAP) Kinases", Journal of Biological Chemistry, vol. 283, pp. 34500-34510, 2008. http://dx.doi.org/10.1074/jbc.M806443200
- V. Levin-Salomon, I. Maayan, L. Avrahami-Moyal, I. Marbach, O. Livnah, and D. Engelberg, "When expressed in yeast, mammalian mitogen-activated protein kinases lose proper regulation and become spontaneously phosphorylated", Biochemical Journal, vol. 417, pp. 331-342, 2008. http://dx.doi.org/10.1042/BJ20081335
- P. Zabrocki, W. Swiatek, E. Sugajska, J.M. Thevelein, S. Wera, and S. Zolnierowicz, "The Saccharomyces cerevisiae type 2A protein phosphatase Pph22p is biochemically different from mammalian PP2A", European Journal of Biochemistry, vol. 269, pp. 3372-3382, 2002. http://dx.doi.org/10.1046/j.1432-1033.2002.02965.x
- M.L. Bailey, B.H. Shilton, C.J. Brandl, and D.W. Litchfield, "The Dual Histidine Motif in the Active Site of Pin1 Has a Structural Rather than Catalytic Role", Biochemistry, vol. 47, pp. 11481-11489, 2008. http://dx.doi.org/10.1021/bi800964q
- H. Kadavath, R.V. Hofele, J. Biernat, S. Kumar, K. Tepper, H. Urlaub, E. Mandelkow, and M. Zweckstetter, "Tau stabilizes microtubules by binding at the interface between tubulin heterodimers", Proceedings of the National Academy of Sciences, vol. 112, pp. 7501-7506, 2015. http://dx.doi.org/10.1073/pnas.1504081112
- S. Tenreiro, M.C. Munder, S. Alberti, and T.F. Outeiro, "Harnessing the power of yeast to unravel the molecular basis of neurodegeneration", Journal of Neurochemistry, vol. 127, pp. 438-452, 2013. http://dx.doi.org/10.1111/jnc.12271
- K. Backhaus, D. Rippert, C.J. Heilmann, A.G. Sorgo, C.G. de Koster, F.M. Klis, R. Rodicio, and J.J. Heinisch, "Mutations in SNF1 complex genes affect yeast cell wall strength", European Journal of Cell Biology, vol. 92, pp. 383-395, 2013. http://dx.doi.org/10.1016/j.ejcb.2014.01.001
- T. Ye, L. Bendrioua, D. Carmena, R. García-Salcedo, P. Dahl, D. Carling, and S. Hohmann, "The mammalian AMP‐activated protein kinase complex mediates glucose regulation of gene expression in the yeast Saccharomyces cerevisiae", FEBS Letters, vol. 588, pp. 2070-2077, 2014. http://dx.doi.org/10.1016/j.febslet.2014.04.039
- J. Rosseels, J. Van den Brande, M. Violet, D. Jacobs, P. Grognet, J. Lopez, I. Huvent, M. Caldara, E. Swinnen, A. Papegaey, R. Caillierez, V. Buée-Scherrer, S. Engelborghs, G. Lippens, M. Colin, L. Buée, M. Galas, E. Vanmechelen, and J. Winderickx, "Tau Monoclonal Antibody Generation Based on Humanized Yeast Models", Journal of Biological Chemistry, vol. 290, pp. 4059-4074, 2015. http://dx.doi.org/10.1074/jbc.M114.627919
- D.F. Tardiff, V. Khurana, C.Y. Chung, and S. Lindquist, "From yeast to patient neurons and back again: Powerful new discovery platforms", Movement Disorders, vol. 29, pp. 1231-1240, 2014. http://dx.doi.org/10.1002/mds.25989
- S. Galanie, K. Thodey, I.J. Trenchard, M. Filsinger Interrante, and C.D. Smolke, "Complete biosynthesis of opioids in yeast", Science, vol. 349, pp. 1095-1100, 2015. http://dx.doi.org/10.1126/science.aac9373
ACKNOWLEDGMENTS
This review is dedicated to the memory of Friedrich („Fritz“) K. Zimmermann, who died in 2014 but whose legacy will live on in generations of German yeast geneticists.
COPYRIGHT
© 2016

Signaling pathways and posttranslational modifications of tau in Alzheimer’s disease: the humanization of yeast cells by Heinisch and Brandt is licensed under a Creative Commons Attribution 4.0 International License.








