
Microreviews:
Microbial Cell, Vol. 1, No. 1, pp. 48 - 50; doi: 10.15698/mic2014.01.124
Stalling autophagy: a new function for Listeria phospholipases

1 Department of Laboratory Medicine and Pathobiology, University of Toronto, M6G 2T6, Toronto, Canada
2 Department of Immunology, University of Toronto, M6G 2T6, Toronto, Canada
Keywords: Listeria, Autophagy, mTOR.
Received originally: 04/12/2013 Received in revised form: 06/12/2013
Accepted: 10/12/2013
Published: 06/01/2014
Correspondence:
Stephen E. Girardin, Department of Laboratory Medicine and Pathobiology, University of Toronto, M6G 2T6, Toronto, Canada stephen.girardin@utoronto.ca
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Ivan Tattoli, Matthew T. Sorbara, Dana J. Philpott and Stephen E. Girardin (2014). Stalling autophagy: a new function for Listeria phospholipases. Microbial Cell 1(1): 48-50.
Listeria monocytogenes is a Gram-positive bacterial pathogen that induces its own uptake in non-phagocytic cells. Following invasion, Listeria escapes from the entry vacuole through the secretion of a pore-forming toxin, listeriolysin O (LLO) that acts to damage and disrupt the vacuole membrane. Listeria then replicates in the cytosol and is able to spread from cell-to-cell using actin-based motility. In addition to LLO, Listeria produces two phospholipase toxins, a phosphatidylinositol-specific phospholipase C (PI-PLC, encoded by plcB) and a broad-range phospholipase C (PC-PLC, encoded by plcA), which contribute to bacterial virulence. It has long been recognized that secretion of PI- and PC-PLC enables the disruption of the double membrane vacuole during cell-to-cell spread, and those phospholipases have also been shown to augment LLO-dependent escape from the entry endosome. However, a specific role for Listeria phospholipases during the cytosolic stage of infection has not been previously reported. In a recent study, we demonstrated that Listeria PI-PLC and PC-PLC contribute to the bacterial escape from autophagy through a mechanism that involves direct inhibition of the autophagic flux in the infected cells [Tattoli et al. EMBO J (2013), 32, 3066-3078].
Autophagy is an essential, highly conserved, catabolic process that plays a critical role in cellular homeostasis and stress response pathways. Autophagy is inhibited by the kinase mTOR, a central cell growth regulator that integrates signals from metabolic cues, such as amino acid and growth factor availability, ATP levels and oxygen tension. Cell starvation triggers mTOR inhibition and autophagy induction, allowing cell to recycle key nutrients, such as amino acids, in order to maintain cellular energy levels. Autophagosomes are structures generated during autophagy, and are formed by the elongation and fusion of smaller isolation membranes (phagophores) into a double-membrane compartment that engulfs cytoplasmic cargo, including dysfunctional cellular components or protein aggregates. The autophagosomes deliver the cargo to lysosomes wherein degradation occurs. This system is also co-opted for degradation of intracellular pathogens, such as Listeria. However, it remained unclear how autophagy is turned on during Listeria infection. Moreover, the mechanisms through which the bacterium escapes autophagy were not fully understood, although a role for the proteins ActA and InlK has been previously shown.
—
We found that Listeria invasion triggered a transient decrease in the phosphorylation of the mTOR complex 1 substrate S6 kinase (S6K), and induced dissociation of mTOR from early/late endosomes. These observations were consistent with an inhibition of the mTOR pathway and coincided with a transient increase in autophagosome targeting to invading Listeria. Upstream of mTOR inhibition, Listeria infection resulted in transient amino acid starvation and activation of the GCN2-eIF2α pathway, which detects and responds to amino acid deficiency. Using LLO- or phospholipase-deficient strains of Listeria, we observed that LLO-dependent damage to the vacuolar membrane was responsible for the pro-autophagy amino acid starvation signals, while recognition of phospholipase activity did not contribute to this early host response. In agreement, our earlier work examining the autophagy response to Shigella and Salmonella infection also revealed a role for amino acid starvation and membrane damage in activating autophagy in infected cells. Together, these results indicate that recognition of pathogen-induced membrane damage leading to amino acid starvation is a general defense mechanism against intracellular pathogens.
—
Following an initial burst of amino acid starvation and autophagy activation, host metabolic pathways in Listeria-infected cells rapidly normalized. This normalization of host metabolic pathways was also observed in Salmonella infected cells, and was necessary and sufficient for the decreased autophagy targeting of Salmonella, since enforced mTOR inhibition using rapamycin increased autophagy targeting and killing of Salmonella. In contrast, stimulation of Listeria infected cells with rapamycin was not sufficient to trigger increased autophagy-dependent killing of the bacteria. These results suggest that, unlike Salmonella, Listeria possesses additional mechanisms to block autophagy once it has escaped to the cytosol.
—
We next examined the mechanism through which Listeria escapes from autophagy once in the cytosol. We noticed that infected cells were characterized by the presence of large granules that contained the membrane damage markers NDP52, Galectin-8, and ubiquitinated proteins, but were not associated with late endosomes or directly targeted to the bacteria. The progressive accumulation of these structures required cytosolic escape, as an LLO-deficient Listeria strain did not induce granule formation. Interestingly, unlike the early transient starvation and autophagy responses induced by LLO-dependent membrane damage and vacuole escape, expression of the phospholipases was required for the accumulation of these granules at late time points of infection. Importantly, while infection with phospholipase-deficient Listeria did not trigger granule formation, the bacteria themselves were more strongly targeted to autophagosomes at late time points of infection. Furthermore, we determined that the formation of these granules was not a result of cell-to-cell spread. Thus, these results indicate that the secreted Listeria phospholipases are likely playing a protective role once the bacteria have escaped to the cytosol (Figure 1). In agreement, production of phospholipases by wild type bacteria was able to protect phospholipase-deficient bacteria from autophagy-dependent killing in co-infection experiments.
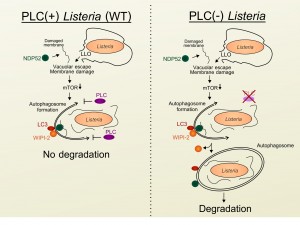 | FIGURE 1: Autophagy stalling by Listeria phospholipases. Schematic representation of the sequential recruitment of autophagy proteins and bacterial targeting in the case of Listeria that do not express phospholipases (PLCs) (left) or wild type (WT) bacteria that express PLCs. LLO, Listeriolysin O. |
In addition to membrane damage markers, we observed that the granules contained the autophagy markers LC3, ATG16L1 and DFCP-1 as well as WIPI-2, a phosphatidylinositol 3-phosphate (PI3P) binding protein that is present on maturing phagophores but is absent from mature autophagosomes (Figure 1). These observations suggested that these phospholipase-triggered granules represented stalled pre-autophagosomal structures. During autophagy induction, PI3P is produced in a Vps34/Beclin1-dependent manner that can be blocked by wortmannin treatment. Since both PI- and PC-PLC are able to cleave phosphatidylinositol (PI), the precursor to PI3P, we investigated whether these molecules affected cellular levels of PI3P. We observed that infection with wild type but not phospholipase-deficient bacteria decreased levels of PI3P, providing a potential mechanism for the phospholipase-dependent manipulation of PI3P pathways. In agreement, short (10 minute) wortmannin treatment of cells infected with phospholipase-deficient bacteria restored granule formation. Furthermore, compared to cells infected with wild type bacteria, autophagy flux was increased in cells infected with phospholipase-deficient Listeria. Collectively, these results indicated that secretion of PI- and PC-PLC into the cytosol by Listeria, interferes with the efficient generation of PI3P resulting in stalling of pre-autophagosome structures, decreased autophagy flux and reduced bacterial targeting to autophagosomes.
—
Following these observations, several important questions remain. First, the biochemical process connecting membrane damage to amino acids starvation during infection remains to be clarified. Second, we determined that Listeria phospholipases inhibit the autophagic flux by reducing PI3P generation. However, the PI3P-binding protein WIPI-2 still accumulated in Listeria phospholipases C-dependent granules. This suggests that pre-autophagosomal structure progression beyond the WIPI-2 step likely requires uncharacterized additional PI3P-dependent events. Finally, the molecular mechanism through which Listeria PLCs down-regulate PI3P levels remains to be characterized.
Overall, this study sheds light onto the mechanisms that were evolved by bacterial pathogens to subvert autophagy. While several previous reports showed that bacteria have designed strategy to disguise from ongoing autophagy, the investigation of how Listeria escapes autophagy through PLC action reveals an interesting mode of action that relies on the direct inhibition of autophagy flux. This concept is similar to the one recently proposed in the case of infections with Legionella pneumophila and Mycobacterium tuberculosis, suggesting that inhibition of autophagy flux by bacterial pathogens is likely a common mechanism evolved by intracellular bacterial pathogens to escape autophagy-mediated clearance.
ACKNOWLEDGMENTS
Research in the laboratory of S.E.G. is supported by grants from the CIHR and NSERC.
COPYRIGHT
© 2014

Stalling autophagy: a new function for Listeria phospholipases by Ivan Tattoli et al. is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.








