
Microreviews:
Microbial Cell, Vol. 4, No. 2, pp. 67 - 68; doi: 10.15698/mic2017.02.559
The tug-of-war over MTOR in Legionella infections

Department of Microbiology and Immunology, Louisiana State University Health Sciences Center – Shreveport, Shreveport, LA 71130.
Keywords: Legionella, macrophage, lipids, lipogenesis, intracellular pathogens, MTOR, PI3K, SREBP.
Received originally: 18/01/2017 Accepted: 23/01/2017
Published: 30/01/2017
Correspondence:
Stanimir S. Ivanov, sivano@lsuhsc.edu
Conflict of interest statement: The author has declared that no competing interests exist.
Please cite this article as: Stanimir S. Ivanov (2017). The tug-of-war over MTOR in Legionella infections. Microbial Cell 4(2): 67-68.
A ruptured bacteria-containing organelle within the cytosol of an infected eukaryotic cell frequently initiates host defense responses that restrict pathogen replication. Therefore, source for lipids must be found to accommodate the organelle membrane expansion required to support bacterial replication. How host cells are manipulated to provide lipids for the expansion of pathogen-occupied organelles is not well understood. By investigating the interaction between macrophages and the human pulmonary pathogen Legionella pneumophila we uncovered that the host metabolic checkpoint kinase Mechanistic target of rapamycin (MTOR) is a central regulator of the pathogen niche expansion program.
–
Previously, we uncovered that mouse bone marrow-derived macrophages (BMMs) infected with pathogenic Legionella suppress MTOR to promote inflammation through a host-driven mechanism. In our recent study (Abshire et al., PLoS Pathogens 2016 Dec 12;12(12):e1006088) we identified the Toll-like Receptor (TLR) adaptor Myeloid differentiation primary response gene 88 (Myd88) as a critical factor mediating the host-induced MTOR suppression in Legionella-infected BMMs. Deletion of Myd88 unmasked an unexpected ability of L. pneumophila to activate and sustain MTOR function even in the absence of growth factors or TLR stimulation. BMMs harboring pathogenic L. pneumophila elicited robust phosphorylation of the ribosomal S6 protein (a downstream substrate of the MTOR pathway) that was sustained for the entire intracellular replication cycle of the bacterium. In sharp contrast, uninfected neighboring cells remained quiescent (Figure 1A and B). Further investigation revealed that one or more protein(s) encoded by L. pneumophila is translocated through the bacterium Type IVb secretion system (T4bSS) directly into the host cytosol to activate MTOR in a phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K)-dependent manner. The Legionella T4bSS manipulates host functions through delivery of ~300 bacterial effector proteins into the host cytosol and is critical for virulence. Thus, it appears that in L. pneumophila-infected macrophages the host and the pathogen are locked in a molecular tug-of-war over MTOR function, where suppression favors the host and activation likely favors the pathogen.
–
The benefit of sustained MTOR activity for Legionella was revealed in part because in MTOR studies cells are generally serum-starved prior to stimulation. Under those conditions, membrane biogenesis requires de novo lipogenesis or lipids recycling through autophagy. Blocking MTOR function in Legionella infections through various approaches destabilized the Legionella-containing vacuole (LCV), led to its premature rupture and the release of bacterial products into the host cytosol, which in turn activated a host cell death response. Culture supplementation with serum but not delipidated serum maintained LCV integrity and blocked the host cell death response implicating a lipids shortage as the likely cause for the loss of LCV integrity under MTOR suppression conditions. Because Legionella efficiently blocks autophagy through LC3 delipidation, the role of MTOR-dependent de novo lipogenesis was investigated. The transcription factors Serum Response Element Binding Protein 1 and 2 (SREBP1/2) are key lipogenesis regulators downstream of MTOR. Legionella infection induced Srebf1 expression as well as SREBP1/2-regulated genes in infected macrophages. Similarly to MTOR suppression, SREBP1/2 inhibition produced a LCV instability phenotype, which was complemented by dietary lipids. Taken together these data indicates that Legionella manipulates the PI3K-MTOR-SREPB1/2 axis to sustain MTOR-dependent lipogenesis to accommodate LCV expansion for optimal intracellular replication.
–
If dietary lipids and de novo lipogenesis are functionally redundant for LCV growth in mammalian macrophages one might wonder why Legionella has retained the capacity to activate MTOR? MTOR anabolic regulation is conserved in unicellular amoebae, the natural hosts for Legionella, which inhabit aquatic environments and lack constant source of dietary lipids. MTOR inhibition reduced Legionella intracellular replication in Acanthamoeba castellanii but only moderately. This observation raises the question if Legionella could manipulate host lipogenesis independent of MTOR and SREBPs or perhaps LCV expansion can be accommodated from pre-existing membranes (Figure 1C). In principle, fusion with other membrane-bound organelles could facilitate LCVs expansion. Indeed, Legionella T4bSS effectors repertoire include proteins that promote recruitment and fusion of the LCV with the early secretory pathway as well as the endoplasmic reticulum. The ER is a major biosynthesis compartment for most cellular lipids and its large membrane reservoir could potentially accommodate some LCV expansion even in the absence of de novo lipogenesis. Considering that we identified a number of Legionella species that replicate intracellularly but do not activate MTOR it would be important to determine the different mechanisms by which LCV compartments expand.
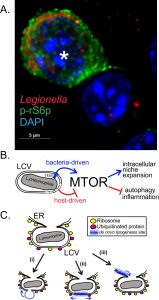 | FIGURE 1: MTOR role in Legionella macrophage infections. (A) A representative micrograph depicting robust MTOR signaling in an infected Myd88-/- BMM (*) but not in an adjacent bystander cell. (B) The balance between multiple regulatory mechanisms dictates MTOR signaling in the Legionella infected macrophages. (C) Putative LCV expansion mechanisms through (i) fusion with membrane-bound organelles, (ii) de novo lipogenesis at the LCV or (iii) through ER-derived membrane infusion. |
Legionella infections could be an excellent model system to decipher how MTOR controls SREBP1/2 and lipogenesis – a key cellular process that is still poorly understood in primary macrophages. Our work points to a rapamycin-insensitive MTOR-dependent mechanism although additional work is needed to elucidate which MTOR complex is involved and the precise mechanism. Host membranes are key features for successful intracellular survival of vacuolar pathogens and we uncovered that subversion of MTOR by L. pneumophila is one mechanism by which the host membrane biogenesis program can be manipulated for niche homeostasis.
ACKNOWLEDGMENTS
The work in my laboratory is supported by a grant from the Louisiana Board of Regents (http://www.regents.la.gov/) (LEQSF(2016-19)-RD-A-15). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
COPYRIGHT
© 2017

The tug-of-war over MTOR in Legionella infections by Stanimir S. Ivanov is licensed under a Creative Commons Attribution 4.0 International License.








