
Research Reports:
Microbial Cell, Vol. 10, No. 9, pp. 195 - 203; doi: doi: 10.15698/mic2023.09.804
Yeast gene KTI13 (alias DPH8) operates in the initiation step of diphthamide synthesis on elongation factor 2
1 Institute of Biology, Division of Microbiology, University of Kassel, Heinrich-Plett-Str. 40, 34132 Kassel, Germany.
2 Roche Pharma Research & Early Development, Large Molecule Research, Roche Innovation Center München, Nonnenwald 2, 82377 Penzberg, Germany.
3 Max-Planck-Institute for Biophysical Chemistry, Am Fassberg 11, 37077 Göttingen, Germany.
4 Environmental Biotechnology, Helmholtz Centre for Environmental Research – UFZ, 04318 Leipzig, Germany.
Keywords: budding yeast; EF2 diphthamide modification; diphtheria toxin; tRNA modification; elongator; tRNase zymocin.
Abbreviations:
KTI – Kluyveromyes lactis toxin insensitive,
FeS – iron-sulfur,
mcm5s2U34 – methoxy-carbonyl-methyl-thio-uridine,
EF2 – elongation factor 2,
DT – diphtheria toxin,
nLC-MS/MS – nano-liquid chromatography tandem mass spectrometry,
YPD – yeast peptone dextrose.
Received originally: 16/05/2023 Received in revised form: 31/07/2023
Accepted: 07/08/2023
Published: 08/08/2023
Correspondence:
Raffael Schaffrath, Institute of Biology, Division of Microbiology, University of Kassel, Heinrich-Plett-Str. 40, 34132 Kassel, Germany; schaffrath@uni-kassel.de
Conflict of interest statement:
KM and UB are employed by and members of Roche Pharma Research & Early Development, and are co-inventors on patent applications that cover assays to de-tect presence or absence of diphthamide. Roche is interested in targeted therapies and diagnostics. All other authors declare no conflict of interest.
Please cite this article as: Meike Arend, Koray Ütkür, Harmen Hawer, Klaus Mayer, Na-mit Ranjan, Lorenz Adrian, Ulrich Brinkmann and Raffael Schaffrath (2023). Yeast gene KTI13 (alias DPH8) operates in the initiation step of diphthamide synthesis on elongation factor 2. Microbial Cell 10(9): 195-203. doi: 10.15698/mic2023.09.804
Abstract
In yeast, Elongator-dependent tRNA modifications are regulated by the Kti11•Kti13 dimer and hijacked for cell killing by zymocin, a tRNase ribotoxin. Kti11 (alias Dph3) also controls modification of elongation factor 2 (EF2) with diphthamide, the target for lethal ADP-ribosylation by diphtheria toxin (DT). Diphthamide formation on EF2 involves four biosynthetic steps encoded by the DPH1-DPH7 network and an ill-defined KTI13 function. On further examining the latter gene in yeast, we found that kti13Δ null-mutants maintain unmodified EF2 able to escape ADP-ribosylation by DT and to survive EF2 inhibition by sordarin, a diphthamide-dependent antifungal. Consistently, mass spectrometry shows kti13Δ cells are blocked in proper formation of amino-carboxyl-propyl-EF2, the first diphthamide pathway intermediate. Thus, apart from their common function in tRNA modification, both Kti11/Dph3 and Kti13 share roles in the initiation step of EF2 modification. We suggest an alias KTI13/DPH8 nomenclature indicating dual-functionality analogous to KTI11/DPH3.
INTRODUCTION
Zymocin is a trimeric (αβγ) chitinase and tRNase toxin complex from Kluyveromyes lactis that kills Saccharomyces cerevisiae cells [1][2]. Expression in S. cerevisiae of its tRNase subunit γ alone (aka γ-toxin) is lethal [3] suggesting subunits α and β mediate zymocin contact with sensitive cells for γ-toxin uptake [2][3]. Accordingly, screens for zymocin survivors identified mutations in non-target (class I) and toxin-target (class II) genes termed KTI (
–
Among KTI loci not coding for Elongator subunits are regulatory genes: KTI11 (aka DPH3), KTI12, KTI13 and KTI14 (aka HRR25) [2][4][15]. Kti12 binds tRNA and supports Elongator phosphorylation by kinase Kti14 [16][17]. Together with Sit4, a phosphatase antagonistic to Kti14, the tRNA modification activity of Elongator likely is phosphoregulated [9][15][18]. Kti11 is a rubredoxin-like electron carrier and dimerizes with Kti13 to effect Elongator-dependent tRNA modifications [18][19]. KTI11 is also allelic with DPH3 [20] and acts in diphthamide decoration of translation elongation factor 2 (EF2), a protein essential for life [21][22][23]. Diphthamide synthesis involves four steps encoded by a network (DPH1–DPH7) that is conserved in eukaryotes [24][25]. The EF2 décor is important for reading frame maintenance during mRNA translation and accurate protein biosynthesis [26][27]. Imbalanced proteostasis as a result of diphthamide deficiency has been attributed to neuropathies and various types of cancer in humans [28]. Of note, diphthamide underlies the human diphtheria disease since it is targeted by corynebacterial diphtheria toxin (DT) for ADP-ribosylation and EF2 inactivation [29] (Fig. 1A).
–
In line with dual roles for tRNA and EF2 modification, Kti11/Dph3 co-purifies with Elongator, EF2 and Dph1•Dph2 [30]. The latter enzyme uses (similar to Elp3) FeS and SAM cofactors for RS chemistry and formation of 3-amino-3-carboxyl-propyl-EF2) (ACP: Fig. 1A), the first diphthamide pathway intermediate [31][32]. Dph3/Kti11 in a dimer with Kti13, donates electrons to the FeS clusters in Dph1•Dph2 and possibly, Elp3 [33][34][35][36], which likely enables proper FeS redox states for RS-based modification chemistry. Whether the dimer feeds into both RS enzymes [18] or limits electron flow to Elongator as suggested [19] is moot. That KTI11/DPH3 and KTI13 gene functions may be related to each modification pathway, is supported by reports showing that both loci genetically interact with the Elongator and EF2 networks [18][22][37][38]. In relation to Kti11/Dph3, however, the precise role of Kti13 and its position within the diphthamide pathway have been less clear. On further studying KTI13 gene function, we found that kti13Δ mutants survive EF2 inhibition by sordarin, a diphthamide-dependent antifungal, and evade ADP-ribosylation of EF2 by DT. Consistently, kti13Δ cells are drastically reduced in ACP formation indicating that proper initiation of the EF2 décor depends on Kti13. This is similar to Kti11/Dph3, which is why we suggest an alias nomenclature: KTI13/DPH8.
–
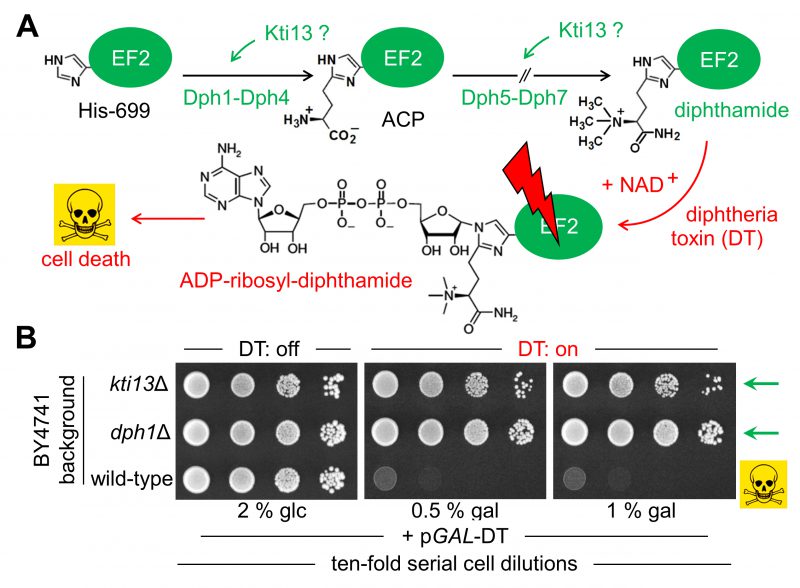 |
FIGURE 1: Potential role of yeast KTI13 in diphthamide modification. (A) Simplified pathway overview [24][25]. Diphthamide synthesis initiates with modifcation of EF2 at His-699 by ACP involving proteins Dph1-Dph4. Subsequential reactions to convert ACP into end product diphthamide entail Dph5-Dph7. Potential Kti13 involvement in the synthesis steps is indicated (‘?’). Diphthamide can be hijacked by diphtheria toxin (DT) for ADP-ribosylation in an NAD+ fashion and induces cell death by EF2 inactivation (skull-crossbones). (B) KTI13 and DPH1 gene deletion strains resist against DT cytotoxicity. Yeast strains carrying pGAL-DT [39], a plasmid for galactose-inducible expression of the lethal ADP-ribosylase domain from DT (see A) were spotted onto medium containing 0.5-1% (w/v) galactose (gal) or 2% (w/v) glucose (glc). Following DT induction, growth inhibition of diphthamide-proficient wild-type is distinguishable from DT resistance of diphthamide-deficient dph1Δ and kti13Δ mutants (green arrows). |
RESULTS AND DISCUSSION
kti13Δ phenotypes diagnostic for a bona fide diphthamide defect
To study Kti13 in more detail (Fig. 1A) we subjected a kti13Δ null-mutant raised in strain BY4741 to DT expression under GAL-promoter control [22][39]. In presence of galactose, DT expression was lethal to wild-type, while kti13Δ cells survived the toxin attack on EF2 (Fig. 1B). The resistance phenotype is robust and similar to the dph1Δ mutant (Fig. 1B), which is blocked in the first step of the diphthamide pathway [21]. Similar to other kti strains or mutants (kti11/dph3Δ, kti12Δ, kti14/hrr25, sit4Δ) lacking Elongator regulators crucial for tRNA modification [2][15][9][40], kti13Δ cells also copied zymocin resistance (Fig. 2A). When we compared growth of this mutant set in the presence of sordarin, a diphthamide-dependent EF2 inhibitor other than DT [22][41], solely kti11/dph3Δ and kti13Δ cells would protect against the antifungal (Fig. 2A). As shown previously, sordarin resistance is a trait diagnostic for failure to initiate or complete EF2 modification with diphthamide [39][41]. Thus, two out of five Elongator regulators tested, apparently share dual-functional roles in tRNA and EF2 modification pathways: Kti11/Dph3 and Kti13.
–
An EF2 pool not modified with diphthamide accumulates in kti13Δ cells
Next, we analyzed protein extracts from the above set of mutants (kti11/dph3Δ, kti12Δ, kti13Δ, kti14/hrr25, sit4Δ) by Western blots (Fig. 2B). We used anti-EF2(pan), an antibody against EF2 regardless of modification, and anti-EF2(no diphthamide) shown to be specific for unmodified EF2 [29][42][43] (Fig. S1). kti13Δ cell extracts produced strong anti-EF2(no diphthamide) Western signals indicative for EF2 species not modified with diphthamide in absence of Kti13 (Fig. 2B). This is a read-out very similar to unmodified EF2 pools from kti11/dph3Δ cells (Fig. 2B), which like other step one mutants (dph1Δ, dph2Δ, dph4Δ) fail to initiate diphthamide synthesis (Fig. 1A) [20][21]. Previously, step one mutants were shown to raise EF2 protein levels, possibly to compensate for diminished EF2 function in absence of diphthamide [25][26]. Here, anti-EF2(pan) Western blots on kti13Δ and kti11/dph3Δ extracts also revealed upregulated EF2 levels (Fig. 2B). Thus, Kti13 and Kti11/Dph3 are diphthamide-related but differ from Kti12, Kti14 and Sit4, which are dispensable for making diphthamide based on anti-EF2(no diphthamide) blots (Fig. 2B). Nonetheless, we observed similar anti-EF2(pan) signals between kti12Δ and kti13Δ cells (Fig. 2B), suggesting an unheard EF2 upregulation under conditions that disturb tRNA (kti12Δ) but not diphthamide modification. In sum, among five known Elongator and tRNA modification regulators, two also contribute to diphthamide modification: Kti11/Dph3 and Kti13. Hence, to go with KTI11/DPH3, we suggest to SGD an alias nomenclature indicating bifunctional nature: KTI13/DPH8.
–
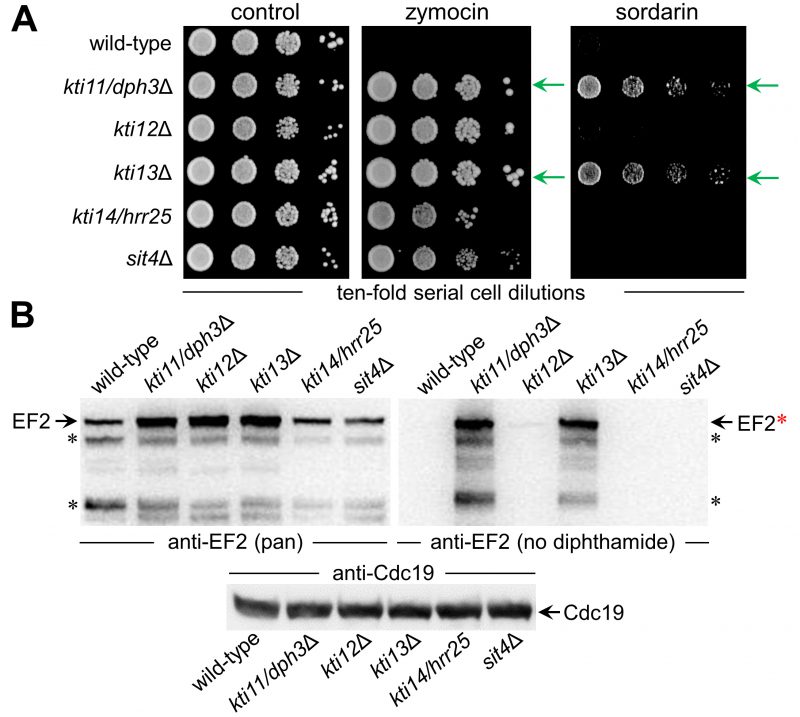 |
FIGURE 2: Among genes involved in Elongator regulation and tRNA modification, KTI11 and KTI13 also function in EF2 modification. (A) Growth assays in response to zymocin (0.02% [v/v]) or sordarin (9 µg/mL) and diagnostic for tRNA or diphthamide modifciation defects, respectively. Dilutions of cells with indicated genotypes were incubated at 30°C for 3 days. Note, that while all ktiΔ and sit4Δ mutants resist growth inhibition by Elongator-dependent tRNase zymocin, only kti11/dph3Δ and kti13Δ cells are protected (green arrows) against diphthamide-dependent EF inhibitor sordarin. (B) Western blot analysis of total cell extracts from strains with genotypes as in A in order to profile their amounts of total EF2 and unmodified EF2 using anti-EF2(pan) (left panel) and anti-EF2(no diphthamide) antibodies (right panel), respectively. Black asterisks (left & right panels) denote EF2 degradation products, the red asterisk indicates full-length unmodified EF2 (right panel). The anti-Cdc19 antibody (bottom panel) was used as loading control. Note the anti-EF2(no diphthamide) Western blot (right panel) detects unmodified EF2 pools for kti11/dph3Δ and kti13Δ cells indicative for diphthamide defects. |
–
kti13Δ cells block proper initiation of EF2 modification with diphtamide
To further examine the position of Kti13 in the diphthamide pathway, we purified His-tagged EF2 from strain TKY675 [44]. Other than the full EF2 gene (EFT1 EFT2) complement of BY4741, TKY675 harbors a double knock-out (eft1Δ eft2Δ) with a single-copy plasmid carrying EFT2-[His]6 [44]. To diagnose diphthamide status in TKY675 prior to EF2 purification, we used DT assays (as above for BY4741). dph1Δ and kti13/dph8Δ mutants survived DT, yet their phenotype was weaker compared to BY4741 counterparts (Fig. 1B) and diminished by increasing DT loads (Fig. 3A). This suggests strain-specific variation due to EF2 copy number effects, a notion supported by Western blots showing significantly reduced EF2 pools and lower (than BY4741) levels of unmodified EF2 in dph1Δ from TKY675 (Fig. S1).
–
Next, we purified His-tagged EF2 from TKY675 in a two-step process coupling immobilized (IMAC: Fig. S2) with size exclusion chromatography (SEC: Fig. S3) for profiling diphthamide modification by nano-liquid chromatography tandem mass spectrometry (nLC-MS/MS) [43]. Previously, yeast, plant and human EF2 were found predominantly diphthamide modified [26][42][43]. In line with this scenario, we hardly detected any unmodified EF2 from total cell extracts of yeast strains BY4741 and TKY675 in anti-EF2(no diphthamide) Western blots (Fig. 2B, Fig. S1). However, upon purification of His-tagged EF2, nLC-MS/MS detected similar amounts of modified and unmodified peptides from TKY675 (wild-type: Fig. 3B). So, in contrast to normal EFT1 EFT2 gene dosage and EF2 levels in BY4741, EF2 purified from TKY675 with single-copy EFT2-[His]6 apparently is not fully modified (Fig. S4). Whether this suggests the affinity-tag on EF2 or gene copy number reduction in TKY675 compromise the modification efficiency of the pathway is unclear. The observed imbalance, however, seems not to be accounted to the His-tag alone based on similar EF2 protein patterns in anti-His versus anti-EF2(pan) Western blots (Fig. S5).
–
Nonetheless, nLC-MS/MS on EF2 purified from the dph1Δ mutant reliably identified an unmodified tryptic peptide with no intermediate or modified variant detectable in TKY675 (Fig. 3B). This is consistent with earlier studies that exclusively identified unmodified EF2 in plant and human dph1Δ cell lines [42][43] and a yeast dph2Δ mutant lacking the Dph1 partner to initiate diphthamide synthesis on EF2 [39][45]. In control purifications from a dph5Δ mutant, which fails to use ACP (Fig. 1A) for formation of methyl-diphthine [39][46], we detected unmodified EF2 (∼35%) and ACP (∼65%) supporting previous data that ACP accumulates when step two of diphthamide pathway is blocked in the absence of Dph5 (Fig. 1A) [46][47]. Importantly, His-tagged EF2 purified from kti13/dph8Δ cells mostly appeared unmodified with minor ACP (∼9%) and low diphthamide (∼1%) amounts (Fig. 3B). Thus, nLC-MS/MS reveals similar profiles among dph1Δ and kti13/dph8Δ mutants strongly suggesting the latter has a step one defect and fails in proper formation of ACP-modified EF2, the first pathway intermediate (Fig. 1A).
–
 |
FIGURE 3: KTI13 is required for proper initiation of diphthamide synthesis on EF2. (A) kti13Δ and dph1Δ mutants in strain TKY675 resist against DT cytotoxicity. The assay was essentially performed as for BY4741 (Fig. 1B). Following galactose-inducible DT expression, wild-type growth inhibition is distinct from DT resistance (green arrows) of diphthamide-deficient mutants (kti13Δ, dph1Δ). (B) Profiling diphthamide modification states on EF2 purified from wild-type, dph1Δ, dph5Δ and kti13Δ cells via nLC-MS/MS. Amounts of modification states were normalized to amounts of unmodified EF2 in dph1Δ (EF2 peptide [%]). kti13Δ contains pools of unmodified EF2 comparable to dph1Δ and drastically reduced ACP levels (∼9%) in relation to dph5Δ (∼65%). (C) ADP-ribosylation (ADPR) assay. Cell extracts from indicated genotypes were incubated with 200 ng DT and biotin-NAD [5 µM] at 25 °C for 1 h. The transfer to EF2 of biotin-ADP-ribose (EF2-ADPR) was detected by Western blot (top panel) using an HRP-streptavidin conjugate recognizing the biotin moiety of the reaction product [26][42]. An anti-Cdc19 Western blot (bottom panel) served as control for sample loading. Note that solely diphthamide-modified EF2 from wild-type cells undergoes detectable ADPR. As has been previously detected in similar assays [29][39], there is an unspecific (n.s.) reaction product of high molecular weight. |
–
Unmodified EF2 from step one kti13Δ mutant escapes ADP-ribosylation by DT
In further support that KTI13/DPH8 deletion copies diphthamide step one mutants are assays using biotinylated NAD+ as ADP-ribosyl donor [26][39] for ADP-ribosylation (ADPR) of EF2 by DT in vitro. Using an HRP-streptavidin conjugate to detect biotin in the ADPR reaction product [42], wild-type EF2 was found to yield robust bio-ADPR-EF2 signals (Fig. 3C). EF2 from dph1Δ or kti13/dph8Δ cells, however, lacked diphthamide-dependent ADPR acceptor activity indicating loss of diphthamide on EF2 evades ADPR by DT (Fig. 3C). These data fully agree with our anti-EF2(no diphthamide) blots showing unmodified EF2 from kti13/dph8Δ and dph1Δ (Fig. 2B; Fig. S1) cells and their DT resistance in vivo (Fig. 1B; Fig. 3A). These are features similar to kti11/dph3Δ cells lacking the electron donor that Kti13/Dph8 dimerizes with to drive Elongator-dependent tRNA modification [18][37]. Whether in analogy, the diphthamide function of Kti11/Dph3 also requires Kti13/Dph8 in the dimer for electron transfer and ACP synthesis by RS enzyme Dph1•Dph2 is plausible given drastically reduced ACP formation in kti13Δ cells (Fig. 3B) and in vivo traits typical of tRNA and EF2 modification loss caused by dimer interface mutations [18][22][37]. However, while electron transfer is essential for Dph1•Dph2 to form ACP in vitro, Kti13/Dph8 is dispensable in theses reconstitution assays [34][37].
CONCLUSION
We show here that apart from its effector role for Elongator-dependent tRNA modification in yeast, Kti13 alias Dph8 also operates in step one of the diphthamide modification pathway (Fig. 4). Although Kti13/Dph8 is important in vivo for EF2 modification by diphthamide, low ACP levels (∼9%) detectable in kti13/dph8Δ cells by MS suggest its presence for the diphthamide pathway to operate is not as catalytically critical as its partner protein Kti11/Dph3 [21][39]. In line with this, previous surveys on the tRNA modification pathway revealed low levels of Elongator activity (∼15%) in kti13Δ but none at all in kti11Δ mutants [9][40]. So residual EF2 and tRNA modification activity in the absence of KTI13/DPH8 suggests an accessory role for gene product Kti13/Dph8. Perhaps it mediates proper electron flow from Kti11/Dph3 to either RS client (Fig. 4) for physiological modification reactions by Dph1•Dph2 and Elongator and thus, helps avoid inappropriate, harmful ones. Alternatively, in the dimer, Kti13/Dph8 may protect its RS clients against oxygen toxicity and FeS cluster damage as recently suggested for yeast and human Dph1•Dph2 [48][49]. Being located upstream of two RS modifiers (Fig. 4) that impact on the accuracy of tRNA decoding and EF2 translocation, a better understanding of how the Kti11•Kti13 (alias Dph3•Dph8) dimer affects mRNA translation is in need [27][50], particularly, in the light of clinically relevant roles for tRNA and EF2 modifications that have recently been shown to emerge in human disease syndromes [51][52].
–
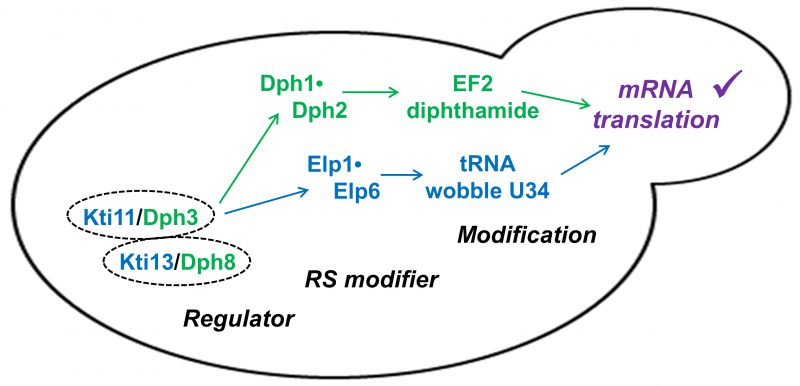 |
FIGURE 4: Kti11•Kti13 dimer (alias Dph3•Dph8), dual modification regulator. The dimer is located upstream of two radical SAM (RS) enzyme complexes (Dph1•Dph2; Elongator: Elp1•Elp6). Its dual regulator roles ensure proper synthesis of diphthamide on EF2 and modification of tRNA anticodon wobble uridine (U34) bases in order to support accurate mRNA translation and de novo protein synthesis [27][50]. |
MATERIALS AND METHODS
Strains, media, growth conditions and assays
S. cerevisiae and K. lactis yeast strains used throughout this study are listed in Table S1. Yeast gene deletion strains were generated based on PCR-mediated protocols using oligonucleotides and gene specific primers (Table S2) with pUG6 plasmid templates [26][39][53]. Strains were grown in complete yeast peptone dextrose (YPD) or minimal synthetic defined (SD) media [54] at 30°C unless otherwise stated. For zymocin response analyses, ten-fold serial cell dilutions of S. cerevisiae tester strains (starting OD600: 1.5) were spotted onto YPD plates lacking or containing 0.02-0.5% (v/v) zymocin. The latter tRNase complex was partially purified from K. lactis killer strain AWJ137 (Table S1) by ultrafiltration [55]. Incubation was for 2-4 days at 30°C. For sordarin assays, yeast cells were cultivated at 30 °C on YPD supplemented with 5-10 μg/mL sordarin produced from Sordaria araneosa (Sigma-Aldrich). DT growth assays involved galactose-inducible expression of the cytotoxic ADP-ribosylase fragment A [29] from DT, using vector pSU9 [39].
–
Analysis of EF2 diphthamide modification status
Diagnosis of EF2 diphthamide modification states in vivo involved Western blots on total yeast cell extracts and antibodies that detect global EF2 pools irrespective of diphthamide modification (anti-EF2[pan]) or specifically recognize unmodified forms of EF2 (anti-EF2[no diphthamide]) [42]. Both antibodies were previously shown to detect human EF2 [42]. As the diphthamide target sequences between human (708-TLHADAIHRGGGQIIPT-724) [42] and yeast (692-TLHADAIHRGGGQIIPT-708) cells are identical [26], anti-EF2(no diphthamide) is suited to differentiate diphthamide modification states of EF2 from S. cerevisiae [26]. Total yeast cell extracts were generated as previously described [56] and protein concentrations determined by the Bradford assay [57]. 8 μl Lämmli samples were subjected to SDS-PAGE (12% [w/v] polyacrylamide) and blotted onto PVDF membranes (Merck/Millipore). These were probed overnight at 4°C with the anti-EF2(pan) anti-EF2(no diphthamide) antibodies [26] and developed with anti-rabbit secondary antibody HRP-conjugate (Dako; working concentration: 1:2000) and Lumi-Light Western blotting substrate (Roche) as previously described [26][42]. Protein loading was controlled in parallel Western blots with anti-Cdc19 antibodies recognizing pyruvate kinase. Diphthamide-dependent ADPR acceptor activity of EF2 in the presence of DT was tested in vitro [58]. The assays used total yeast extracts and biotinylated NAD+ as ADP-ribosyl donor for DT essentially as previously described with human and yeast EF2 resources [58][59].
–
Two-step purification of His-affinity tagged EF2 by IMAC and SEC
His-tagged EF2 from strain TKY675 carrying EFT2-[His]6 on pTKB612 (Table S1) was detected with anti-(His)6 antibodies (Santa Cruz Biotechnology, USA) in Western blots. Purification by IMAC and SEC used 5 ml HisTrap columns (GE Healthcare, Chicago, USA) (Fig. S2, S3). For detailed IMAC and SEC protocols including modifications from the one originally described [44], see Supplemental Material.
–
Detection of EF2 diphthamide modification states by mass spectrometry
Isolated EF2 proteins from the various TKY675 backgrounds (wild-type, dph1Δ, dph5Δ and kti13/dph8Δ) were analyzed via nLC-MS/MS to determine their diphthamide modification states in accordance with an earlier description for EF2 modification analysis from Arabidopsis thaliana [43]. Yeast proteins were separated by SDS-PAGE (7.5% [w/v] polyacrylamide), stained with Coomassie Blue, and excised as bands from the gel. Disulfides were reduced with dithionite and cysteine residues were alkylated with iodoacetamide, followed by trypsin digestion of proteins overnight, all within the gel piece as described [60]. Trypsin-digested fragments were eluted from the gel pieces and desalted using ZipTips [61] before analysis by nLC-MS/MS on a Thermo Orbitrap Fusion mass spectrometer (ThermoFisher) with injection via an electrospray ion source (Tri-Versa NanoMate, Advion). Acquisition of mass spectra was done at a resolution of 120,000 for MS1 scans and 60,000 for MS2 scans with operation parameters described in detail elsewhere [61]. Diphthamide-modified (C81H137N25O23), ACP-modified (C78H130N24O24) and diphthamide-unmodified (C74H123N23O22) precursor masses of 1829,03, 1786,97, and 1685,92, respectively, of target peptide 686-VNILDVTLHADAI
REFERENCES
- Butler AR, O'Donnell RW, Martin VJ, Gooday GW, and Stark MJ (1991). Kluyveromyces lactis toxin has an essential chitinase activity. Eur J Biochem 199: 483–488. 10.1111/j.1432-1033.1991.tb16147.x
- Jablonowski D, and Schaffrath R (2007). Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem Soc Trans 35: 1533–1537. 10.1042/BST0351533
- Stark MJ, Boyd, A, Mileham AJ, and Romanos, MA (1990). The plasmid-encoded killer system of Kluyveromyces lactis: a review. Yeast 6: 1–29. 10.1002/yea.320060102
- Butler AR, White JH, Folawiyo Y, Edlin A, Gardiner D, and Stark MJ (1994). Two Saccharomyces cerevisiae genes which control sensitivity to G1 arrest induced by Kluyveromyces lactis toxin. Mol Cell Biol 14: 6306–6316. 10.1128/mcb.14.9.6306-6316.1994
- Jablonowski D, Fichtner L, Martin VJ, Klassen R, Meinhardt F, Stark MJ, and Schaffrath, R (2001). Saccharomyces cerevisiae cell wall chitin, the Kluyveromyces lactis zymocin receptor. Yeast 18: 1285–1299. 10.1002/yea.776
- Mehlgarten C, and Schaffrath R (2004). After chitin-docking, toxicity of Kluyveromyces lactis zymocin requires Saccharomyces cerevisiae plasma membrane H+-ATPase. Cell Microbiol 6: 569–580. 10.1111/j.1462-5822.2004.00383.x
- Zink S, Mehlgarten C, Kitamoto HK, Nagase J, Jablonowski D, Dickson RC, Stark MJ, and Schaffrath, R (2005). M(IP)2C, the major yeast plasma membrane sphingolipid, governs toxicity of Kluyveromyces lactis zymocin. Eukaryot Cell 4: 879–889. 10.1128/EC.45.879-889.2005
- Frohloff F, Fichtner L, Jablonowski D, Breunig KD, and Schaffrath, R (2001). Saccharomyces cerevisiae Elongator mutations confer resistance to the Kluyveromyces lactis zymocin. EMBO J 20: 1993–2003. 10.1093/emboj/20.8.1993
- Huang B, Johansson MJ, and Bystrom AS (2005). An early step in wobble uridine tRNA modification requires the Elongator complex. RNA 11: 424–436. 10.1261/rna.7247705
- Paraskevopoulou C, Fairhurst SA, Lowe DJ, Brick P, and Onesti S (2006). The Elongator subunit Elp3 contains a Fe4S4 cluster and binds S-adenosylmethionine. Mol Microbiol 59: 795–806. 10.1111/j.1365-2958.2005.04989.x
- Lin TY, Abbassi NEH, Zakrzewski K, Chramiec-Głąbik A, Jemioła-Rzemińska M, Różycki J, and Glatt S (2019). The Elongator subunit Elp3 is a non-canonical tRNA acetyltransferase. Nat Commun 10: 625. 10.1038/s41467-019-08579-2
- Abbassi NE, Biela A, Glatt S, and Lin TY (2020). How Elongator acetylates tRNA bases. Int J Mol Sci 21: 8209. 10.3390/ijms21218209
- Lu J, Huang B, Esberg A, Johansson MJ, and Byström AS (2005). The Kluyveromyces lactis gamma-toxin targets tRNA anticodons. RNA 11: 1648–1654. 10.1261/rna.2172105
- Jablonowski D, Zink S, Mehlgarten C, Daum G, and Schaffrath R (2006). tRNAGlu wobble uridine methylation by Trm9 identifies Elongator's key role for zymocin-induced cell death in yeast. Mol Microbiol 59: 677–688. 10.1111/j.1365-2958.2005.04972.x
- Schaffrath R, and Leidel SA (2017). Wobble uridine modifications – a reason to live, a reason to die?! RNA Biol 14: 1209–1222. 10.1080/15476286.2017.1295204
- Krutyhołowa R, Hammermeister A, Zabel R, Abdel-Fattah W, Reinhardt-Tews A, Helm M, Stark MJ, Breunig KD, Schaffrath R, and Glatt S. (2019) Kti12, a PSTK-like tRNA dependent ATPase essential for tRNA modification by Elongator. Nucleic Acids Res 47: 4814–4830. 10.1093/nar/gkz190
- Abdel-Fattah W, Jablonowski D, Di Santo R, Scheidt V, Hammermeister A, ten Have SM, Thüring KL, Helm M, Schaffrath, R, and Stark MJ (2015). Phosphorylation of Elp1 by Hrr25 is required for Elongator-dependent tRNA modification in yeast. PLoS Genet 11: e1004931. 10.1371/journal.pgen.1004931
- Glatt S, Zabel R, Vonkova I, Kumar A, Netz DJ, Pierik AJ, Rybin V, Lill R, Gavin AC, Balbach J, Breuing KD, and Müller CW (2015). Structure of the Kti11/Kti13 heterodimer and its double role in modification of tRNA and eukaryotic elongation factor 2. Structure 23: 149–160. 10.1016/j.str.2014.11.008
- Kolaj-Robin O, McEwen AG, Cavarelli J, and Séraphin B (2015). Structure of the Elongator cofactor complex Kti11/Kti13 provides insight into the role of Kti13 in Elongator-dependent tRNA modification. FEBS J 282: 819–833. 10.1111/febs.13199
- Liu S, and Leppla SH (2003). Retroviral insertional mutagenesis identifies a small protein required for synthesis of diphthamide, the target of bacterial ADP-ribosylating toxins. Mol Cell 12: 603–613. 10.1016/j.molcel.2003.08.003
- Liu S, Milne GT, Kuremsky JG, Fink GR, and Leppla SH (2004). Identification of the proteins required for biosynthesis of diphthamide, the target of bacterial ADP-ribosylating toxins on translation elongation factor 2. Mol Cell Biol 24: 9487–9497. 10.1128/MCB.24.21.9487-9497.2004
- Bär C, Zabel R, Liu S, Stark MJ, and Schaffrath R (2008). A versatile partner of eukaryotic protein complexes that is involved in multiple biological processes: Kti11/Dph3. Mol Microbiol 69: 1221–1233. 10.1111/j.1365-2958.2008.06350.x
- Jørgensen R, Merrill AR, and Andersen GR (2006). The life and death of translation elongation factor 2. Biochem Soc Trans 34: 1–6. 10.1042/BST20060001
- Su X, Lin Z, and Lin H (2013). The biosynthesis and biological function of diphthamide. Crit Rev Biochem Mol Biol 48: 515–521. 10.3109/10409238.2013.831023
- Schaffrath R, Abdel-Fattah W, Klassen, R, and Stark MJ (2014). The diphthamide modification pathway from Saccharomyces cerevisiae – Revisited. Mol Microbiol 94: 1213–1226. 10.1111/mmi.12845
- Hawer H, Ütkür K, Arend M, Mayer K, Adrian L, Brinkmann U, Schaffrath R (2018). Importance of diphthamide modified EF2 for translational accuracy and competitive cell growth in yeast. PLoS One 13: e0205870. 10.1371/journal.pone.0205870
- Djumagulov M, Demeshkina N, Jenner L, Rozov A, Yusupov M, Yusupova G (2021). Accuracy mechanism of eukaryotic ribosome translocation. Nature 600: 543–546. 10.1038/s41586-021-04131-9
- Hawer H, Hammermeister A, Ravichandran KE, Glatt S, Schaffrath R, Klassen R (2019). Roles of Elongator dependent tRNA modification pathways in neurodegeneration and cancer. Genes 10: 19. 10.3390/genes10010019
- Uthman S, Liu S, Giorgini F, Stark MJ, Costanzo M, and Schaffrath R (2012). Diphtheria disease and genes involved in formation of diphthamide, key effector of the diphtheria toxin. In: Insight and Control of Infectious Disease in Global Scenario, Kumar R (ed), INTECH OAP, pp 333–356. 10.5772/31680
- Fichtner L, Jablonowski D, Schierhorn A, Kitamoto HK, Stark MJ, and Schaffrath R (2003). Elongator's toxin-target (TOT) function is NLS dependent and suppressed by post-translational modification. Mol Microbiol 49: 1297–1307. 10.1046/j.1365-2958.2003.03632.x
- Zhang Y, Zhu X, Torelli AT, Lee M, Dzikovski B, Koralewski RM, Wang E, Freed J, Krebs C, Ealick SE, and Lin H (2010). Diphthamide biosynthesis requires an organic radical generated by an iron-sulphur enzyme. Nature 465: 891–896. 10.1038/nature09138
- Dong M, Dando EE, Kotliar I, Su X, Dzikovski B, Freed JH, and Lin H (2019). The asymmetric function of Dph1-Dph2 heterodimer in diphthamide biosynthesis. J Biol Inorg Chem 24: 777–782. 10.1007/s00775-019-01702-0
- Sun J, Zhang J, Wu F, Xu C, Li S, Zhao W, Wu Z, Wu J, Zhou CZ, and Shi Y (2005). Solution structure of Kti11p from Saccharomyces cerevisiae reveals a novel zinc-binding module. Biochemistry 44: 8801–8809. 10.1021/bi0504714
- Dong M, Su X, Dzikovski B, Dando EE, Zhu X, Du J, Freed JH, and Lin H (2014). Dph3 is an electron donor for Dph1-Dph2 in the first step of eukaryotic diphthamide biosynthesis. J Am Chem Soc 136: 1754–1757. 10.1021/ja4118957
- Zhang Y, Su D, Dzikovski B, Majer SH, Coleman R, Chandrasekaran S, Fenwick MK, Crane BR, Lancaster KM, Freed JH, and Lin H (2021). Dph3 enables aerobic diphthamide biosynthesis by donating one iron atom to transform a [3Fe-4S] to a [4Fe-4S] cluster in Dph1-Dph2. J Am Chem Soc 143: 9314–9319. 10.1021/jacs.1c03956
- Lin Z, Dong M, Zhang Y, Lee EA, and Lin H (2016). Cbr1 is a Dph3 reductase required for the tRNA wobble uridine modification. Nat Chem Biol 12: 995–997. 10.1038/nchembio.2190
- Zabel R, Bär C, Mehlgarten C, and Schaffrath, R (2008). Yeast-tubulin suppressor Ats1/Kti13 relates to the Elongator complex and interacts with Elongator partner protein Kti11. Mol Microbiol 69: 175–187. 10.1111/j.1365-2958.2008.06273.x
- Fichtner L, and Schaffrath R (2002). KTI11 and KTI13, Saccharomyces cerevisiae genes controlling sensitivity to G1 arrest induced by Kluyveromyces lactis zymocin. Mol Microbiol 44: 865–875. 10.1046/j.1365-2958.2002.02928.x
- Uthman S, Bär C, Scheidt V, Liu S, ten Have S, Giorgini F, Stark MJ, and Schaffrath R (2013). The amidation step of diphthamide biosynthesis in yeast requires DPH6, a gene identified through mining the DPH1–DPH5 interaction network. PLoS Genet 9: e1003334. 10.1371/journal.pgen.1003334
- Huang B, Lu J, and Byström AS (2008). A genome-wide screen identifies genes required for formation of the wobble nucleoside 5-methoxycarbonylmethyl-2-thiouridine in Saccharomyces cerevisiae. RNA 14: 2183–2194. 10.1261/rna.1184108
- Shao Y, Molestak E, Su W, Stankevič M, Tchórzewski M (2022). Sordarin – an anti-fungal antibiotic with a unique modus operandi. Br J Pharmacol 179: 1125–1145. 10.1111/bph.15724
- Stahl S, da Silva Mateus Seidl AR, Ducret A, Kux van Geijtenbeek S, Michel S, Racek T, Birzele F, Haas AK, Rueger R, Gerg M, Niederfellner G, Pastan I, and Brinkmann U (2015). Loss of diphthamide pre-activates NF-kappaB and death receptor pathways and renders MCF7 cells hypersensitive to tumor necrosis factor. Proc Natl Acad Sci USA 112: 10732–10737. 10.1073/pnas.1512863112
- Zhang H, Quintana J, Ütkür K, Adrian L, Hawer H, Mayer K, Gong X, Castanedo L, Schulten A, Janina N, Peters M, Wirtz M, Brinkmann U, Schaffrath R, and Krämer U (2022). Translational fidelity and growth of Arabidopsis require stress-sensitive diphthamide biosynthesis. Nat Comm 13: 4009. 10.1038/s41467-022-31712-7
- Jørgensen R, Carr-Schmid A, Ortiz PA, Kinzy TG, and Andersen, GR (2002). Purification and crystallization of the yeast elongation factor eEF2. Acta Crystallogr D Biol Crystallogr 58: 712–715. 10.1107/s0907444902003001
- Ortiz PA, Ulloque R, Kihara GK, Zheng H, and Kinzy TG (2006). Translation elongation factor 2 anticodon mimicry domain mutants affect fidelity and diphtheria toxin resistance. J Biol Chem 281: 32639–32648. 10.1074/jbc.M607076200
- Mattheakis LC, Shen WH, and Collier RJ (1992). DPH5, a methyltransferase gene required for diphthamide biosynthesis in Saccharomyces cerevisiae. Mol Cell Biol 12: 4026–4037. 10.1128/mcb.12.9.4026-4037.1992
- Lin Z, Su X, Chen W, Ci B, Zhang S, and Lin H (2014). Dph7 catalyzes a previously unknown demethylation step in diphthamide biosynthesis. J Am Chem Soc 136: 6179–6182. 10.1021/ja5009272
- Zhang Y, Su D, Dzikovski B, Majer SH, Coleman R, Chandrasekaran S, Fenwick MK, Crane BR, Lancaster KM, Freed JH, and Lin H (2021). Dph3 enables aerobic diphthamide biosynthesis by donating one iron atom to transform a [3Fe-4S] to a [4Fe-4S] cluster in Dph1-Dph2. J Am Chem Soc 143: 9314–9319. 10.1021/jacs.1c03956
- Baik AH, Haribowo AG, Chen X, Queliconi BB, Barrios AM, Garg A, Maishan M, Campos AR, Matthay MA, and Jain IH (2023). Oxygen toxicity causes cyclic damage by destabilizing specific Fe-S cluster-containing protein complexes. Mol Cell 83: 942–960.e9. 10.1016/j.molcel.2023.02.013
- Nedialkova DD, and Leidel SA (2015). Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161: 1606–1618. 10.1016/j.cell.2015.05.022
- Hawer H, Mendelsohn BA, Mayer K, Kung A, Malhotra A, Tuupanen S, Schleit J, Brinkmann U, and Schaffrath R (2020). Diphthamide-deficiency syndrome: a novel human developmental disorder and ribosomopathy. Eur J Hum Genet 28: 1497–1508. 10.1038/s41431-020-0668-y
- Gaik M, Kojic M, Wainwright BJ, and Glatt S (2023). Elongator and the role of its subcomplexes in human diseases. EMBO Mol Med 15: e16418. 10.15252/emmm.202216418
- Gueldener U, Heinisch J, Koehler GJ, Voss D, and Hegemann JH (2002). A second set of loxP marker cassettes for Cre-mediated multiple gene knockouts in budding yeast. Nucleic Acids Res 30: e23. 10.1093/nar/30.6.e23
- Sherman F (2002). Getting started with yeast. Methods Enzymol 350: 3–41. 10.1016/s0076-6879(02)50954-x
- Klassen R, Wemhoff S, Krause J, and Meinhardt F (2011). DNA repair defects sensitize cells to anticodon nuclease yeast killer toxins. Mol Genet Genomics 285: 185–195. 10.1007/s00438-010-0597-5
- Zachariae W, Shin TH, Galova M, Obermaier B, abd Nasmyth K (1996). Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. Science 274: 201–1204. 10.1126/science.274.5290.1201
- Bradford MM (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Chem 72: 248–254. 10.1006/abio.1976.9999
- Mayer K, Schröder A, Schnitger J, Stahl S, and Brinkmann U (2017). Influence of DPH1 and DPH5 protein variants on the synthesis of diphthamide, the target of ADPRibosylating toxins. Toxins 9: 78. 10.3390/toxins9030078
- Shankar SP, Grimsrud K, Lanoue L, Egense A, Willis B, Hörberg J, Alabdi L, Mayer K, Ütkür K, Monaghan KG, Krier J, Stoler J, Alnemer M, Shankar PR, Schaffrath R, Alkuraya FS, Brinkmann U, Eriksson LA, Lloyd K, Rauen KA, and Undiagnosed Diseases Network (2022). A novel DPH5-related diphthamide-deficiency syndrome causing embryonic lethality or profound neurodevelopmental disorder. Genet Med 24: 2207. 10.1016/j.gim.2022.07.021
- Kublik A, Deobald D, Hartwig S, Schiffmann CL, Andrades A, von Bergen M, Sawers RG, and Adrian L (2016). Identification of a multi-protein reductive dehalogenase complex in Dehalococcoides mccartyi strain CBDB1 suggests a protein dependent respiratory electron transport chain obviating quinone involvement. Environ Microbiol 18: 3044–3056. 10.1111/1462-2920.13200
- Seidel K, Kuhnert J, and Adrian L (2018). The complexome of Dehalococcoides mccartyi reveals its organohalide respiration-complex is modular. Front Microbiol 9: 1130. 10.3389/fmicb.2018.01130
–
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We thank Tessa Hübner (MPI, Göttingen, Germany) and Benjamin Scheer (UFZ, Leipzig, Germany) for assistance with EF2 purification and nLC-MS/MS, Dr Roland Klassen (Kassel University, Germany) for critical reading of the manuscript and Prof Jeremy Thorner (University of California, Berkeley, USA) for kindly donating anti-Cdc19 antibodies. The work was supported by DFG (Bonn, Ger-many) Priority Program 1927 Iron-Sulfur for Life to LA (AD178/7-1) and RS (SCHA750/21-1) and by a Diphthamide Pilotgrant to RS (#2887) from ZFF (Kassel University, Germany).
COPYRIGHT
© 2023

Yeast gene KTI13 (alias DPH8) operates in the initiation step of diphthamide synthesis on elongation factor 2 by Arend et al. is licensed under a Creative Commons Attribution 4.0 International License.








