
Reviews:
Microbial Cell, Vol. 3, No. 7, pp. 263 - 274; doi: 10.15698/mic2016.07.510
Evidence for the hallmarks of human aging in replicatively aging yeast

European Research Institute for the Biology of Ageing, University of Groningen, University Medical Centre Groningen, Antonius Deusinglaan 1, 9713 AV, Groningen, The Netherlands.
Keywords: hallmarks of aging, human, replicative aging, yeast.
Abbreviations:
ERC – extrachromosomal rDNA circle,
GO – gene ontology,
NPC – nuclear pore complex,
RLS – replicative lifespan,
ROS – reactive oxygen species.
Received originally: 17/02/2016 Received in revised form: 19/04/2016
Accepted: 20/04/2016
Published: 20/06/2016
Correspondence:
Liesbeth Veenhoff, European Research Institute for the Biology of Ageing, University of Groningen, University Medical Centre Groningen, Antonius De-usinglaan 1, 9713 AV, Groningen, The Netherlands. l.m.veenhoff@rug.nl
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Georges E. Janssens, Liesbeth M. Veenhoff (2016). Evidence for the hallmarks of human aging in replicatively aging yeast. Microbial Cell 3(7): 263-274.
Abstract
Recently, efforts have been made to characterize the hallmarks that accompany and contribute to the phenomenon of aging, as most relevant for humans [1]. Remarkably, studying the finite lifespan of the single cell eukaryote budding yeast (recently reviewed in [2] and [3]) has been paramount for our understanding of aging. Here, we compile observations from literature over the past decades of research on replicatively aging yeast to highlight how the hallmarks of aging in humans are present in yeast. We find strong evidence for the majority of these, and summarize how yeast aging is especially characterized by the hallmarks of genomic instability, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, and mitochondrial dysfunction.
INTRODUCTION
The field of the biology of aging has benefited immensely from studying the finite lifespan of the budding yeast Saccharomyces cerevisiae (recently reviewed in [2] and [3]). Aging can be assessed in multiple ways in this single-celled eukaryote, the most common of which are by measuring the limited number of replications it can undergo [4], or by measuring the chronological time it can spend in a non-dividing state before losing viability [5]. Termed the replicative lifespan (RLS) and chronological lifespan (CLS), respectively, overlap and differences exist between the factors affecting lifespan in the two models of aging [6]. Our review here will focus on the RLS model of aging.
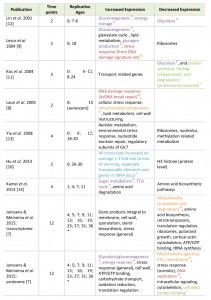 | TABLE 1. Overview of system wide gene expression studies in replicatively aging yeast. Publications covering either transcriptomes (first 8 rows) or proteome (last row) of wild-type aging yeast. General terms of increasing or decreasing changes are taken as described in the original publications. *Ages inferred from hours of cultivation, assuming a two-hour division time. Color code and superscripts associate changes reported in yeast relative to the hallmarks of human aging as seen in Figure 1 and described in the text; ‘G’ genomic instability (red), ‘E’ epigenetic alterations (blue), ‘P’ loss of proteostasis (green), ‘D’ deregulated nutrient sensing (purple), ‘M’ mitochondrial dysfunction (orange). [7][8][9][10][11][12][13][14] |
–
Recently, an effort has been made to characterize the hallmarks that accompany aging [1] as most applicable to humans. Defined as the time-dependent functional decline affecting living organisms, nine hallmarks were designated as being contributors to the aging process [1]. These hallmarks are: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [1]. The purpose of our review is to place the aging related changes occurring in yeast within the context of these hallmarks of aging from higher eukaryotes, by collecting and organizing relevant literature (Table 1, Figure 1), and exploring pathways that change with age from proteome data resulting from our recent survey of aging yeast [7] (Figure 2). A yeast cell begins its life emerging as a bud from a mother cell. It has had its age reset, rejuvenated when compared to its mother. Soon after separation from the mother cell, it begins to produce daughters of its own. Now also a ‘mother,’ it will undergo a limited number of divisions, which is a model for aging of mitotically active cells in higher eukaryotes [2]. The entirety of this process essentially reflects the hallmarks of aging for stem cell exhaustion and to some degree [8][9] cellular senescence in humans. These hallmarks however are intended to capture the decline in regenerative potential of tissues that occurs with aging [1], considered to be in fact partially affected by cell extrinsic factors [10], and we will not further elaborate on these. Several other hallmarks of human aging will not be discussed in detail here, because they have limited relevance for natural aging in yeast, or they have not been robustly implicated in yeast aging. For example, though telomeres have been implicated in yeast aging ([11][12], Table 1), no significant telomere attrition occurs while the mother cell is replicating [13]. Meanwhile, for altered intercellular communication, signs are present of its involvement relevant to yeast aging but only in limited form [14][15][16]. Rather, much more compelling evidence exists for the hallmarks of genomic instability, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, and mitochondrial dysfunction in the yeast replicative lifespan (Figure 1), which are described in greater detail below.
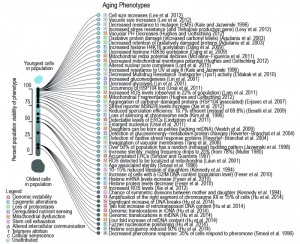 | FIGURE 1: Phenotypes occurring in the yeast replicative lifespan. Observations collected from primary literature as discussed herein and as associated to a relevant hallmark of aging (symbol in legend). Cited references are often not the first to observe the phenotype but are selected based on the earliest most precise report of the timing within the RLS (i.e. while [23][24] described aged cells to be larger than young, [25] clearly observed this increase in size for a specific age, namely from the beginning on, when 100% of the population was still viable). Percentage of viability, at which age-related phenotype occurs in the population, was either directly taken from the primary literature source, or inferred based on the age of the yeast used in the publication, its average lifespan (if provided), or the average lifespan estimated from a compiled lifespan curve. |
GENOMIC INSTABILITY
Genomic instability, defined simply as unintended alterations occurring in the genome [17], has been hypothesized to be central to aging for over half a century, as exemplified by the proposal of the Somatic Mutation Theory of Aging [18][19]. This has been supported by evidence that the integrity of DNA is constantly challenged by chemical, physical, and biological agents that react with DNA [20], and that somatic mutations accumulate in cells of aging humans and model organisms [21]. Furthermore, the fact that defects in the DNA repair machinery cause diseases of accelerated aging such as Werner and Bloom syndromes [22], has earned genomic instability the title of being a hallmark of aging.
 | FIGURE 2: Age-related changes in Gene Ontology Processes. Gene ontology process categories taken from GO Slim lists of yeastgenome.org. Relevant Hallmarks of Aging are listed next to terms, as discussed in the text, same legend as Figure 1. Plot is comparison of old (~72 hours of aging with roughly 42% viability of the population) versus young (~8 hours of aging with nearly 100% viability of the population) protein-level changes from the [7] dataset. Data expressed in terms of fold change on a log2 scale. Red dashed line indicates two fold change. Blue solid line zero change. Numbers of proteins measured per GO term are listed out of total proteins in category. Distributions of fold changes per GO term expressed as horizontal box-and-whisker plots. The thick black line within the box is the median of the data, the box extends to the upper and lower quartile of the dataset (i.e. to include 25% of the data above and below the median, respectively), whiskers (dashed lines) represent up to 1.5 times the upper or lower quartiles and circles represent outliers. |
–
In yeast, genomic instability has been a subject of extensive research due to its relevance to cancer [23]. In relation to aging, SGS1, which encodes a RecQ helicase and is homologous to the genes mutated in Werner and Bloom syndromes in humans, shortens lifespan drastically when removed [24]. Likewise, removing other proteins in the DNA damage response pathway, such as the 5′ to 3′ exonuclease and 5′ flap endonuclease Rad27 and the single-stranded DNA-dependent ATPase, ATP-dependent nuclease, and helicase Dna2, has been shown to result in decreased lifespan for the cell [25]. At the transcriptional level, a signature of DNA damage has been described to occur during aging [26] (Table 1), indicating the presence of insults to the genome, and furthermore, diploid yeast have been shown to lose heterozygosity with aging [27], likely due to compromised maintenance of genome integrity [27][28].
–
Although responses to DNA instability seem to exist in aging yeast cells, it has been widely accepted that the accumulation of mutations is not a cause of aging for yeast [2]. This is clear from the observation that while daughters of advanced age mothers are born aged, their granddaughters are born rejuvenated, indicating that the genome has remained intact and operable [29]. Therefore, observations of genetic changes such as increased transposable element activity and amplification of chromosome arms are thought only to occur in extreme late life [30]. Through recent genome sequencing searching for mutations in aging yeast, evidence was found that the integrity of the genome is preserved during replication, and the accumulation of mutations is not considered to be a cause of aging in yeast [31].
–
A more relevant interpretation of aging related genomic instability in yeast could be related to extrachromosomal rDNA circles (ERCs) [32]. ERCs are circles of DNA from the rDNA region of the genome, which may loop out by homologous recombination after attempts occur to repair a nearby double strand break [32]. Once formed, the presence of an origin of replication on the ERC causes its replication during S-phase of the cell cycle [32]. With a biased retention of ERCs in the mother cell, these circles of DNA accumulate with replicative age [32]. While the exact mechanism of how ERCs are asymmetrically retained in aging cells remains debated (see [2] for a discussion), what is clear is their influence on aging: reducing their formation results in increased lifespan while artificially increasing their formation results in shorter lifespan [32][33]. Indeed, accumulation of any self-replicating circular DNA has been shown to decrease lifespan in yeast [34]. Furthermore, it has been proposed that genomic stability in the rDNA locus is the mechanism by which mutants that decrease ERC formation, increase lifespan, supporting an ‘rDNA-theory of aging’ [35][36][37][38][39]. Finally, it has been shown that rDNA undergoes genomic translocations with aging [30], a sign of genomic instability.
–
It has been proposed that the accumulation of ERCs burden the normal replication machinery over time, compromising the cell’s ability to replicate genomic DNA, thereby limiting lifespan [32]. This has been supported by the observation that a yeast mutant over-amplifying 2μ DNA circles possesses a relatively longer S-phase [40]. Though the exact mechanism by which the instability at the rDNA locus limits lifespan is not fully known, it is well established that ERCs are a result of instability in the genome [36][37] and it is therefore clear that the hallmark of genomic instability is a potent factor in the replicative lifespan of yeast.
EPIGENETIC ALTERATIONS
Epigenetic alterations of the genome, such as those of DNA methylation patterns, posttranslational histone modifications, and chromatin remodeling, have been termed a hallmark of aging [1]. For example in mammals, increased trimethylation of histones H4K20 (seen in rat liver [41]) and H3K4 (seen in neurons from human prefrontal cortex [42]), have been reported to occur (reviewed in [43] and [44] along with epigenetic marks in model organisms). Adding to this, modification of certain components of histone modifying complexes extends lifespan in worms and flies [45][46][47]. Perhaps most convincingly for its relevance to humans, the DNA methylation state of blood cells was recently shown to have predictive ability on all-cause mortality in later life [48][49].
–
In yeast, epigenetic alterations have been shown to have a strong influence on lifespan, especially when considering changes in histones. Perhaps most notably, the histone deacetylase Sir2 has been shown to have lifespan extending effects when overexpressed in yeast [50], as in worms and flies [51]. Furthermore, increased histone H4K16 acetylation and decreased H3K56 acetylation has been observed to occur with aging [52]. This age-related increase of H4K16 acetylation is thought to be related to the natural decline in levels of Sir2 that occurs with aging [52], which is responsible for deacetylation of H4K16 [53]. Additionally, specific gene deletions in the SAGA/SLIK (Spt-Ada-Gcn5-Acetyltransferase)/(SAGA-Like) histone deubiquitinase modules have been shown to increase lifespan in a Sir2-dependent manner [54]. Furthermore, histone protein levels themselves have been shown to go down with aging [55]. This decline in abundance of histones clearly has a direct effect on the lifespan of the cells, since counteracting this loss of histone levels has been shown to increase lifespan in a manner seemingly independent from other longevity pathways [55].
–
Overall, we find evidence for aging-related changes in epigenetic control systems in yeast (Figure 1, Figure 2). This hallmark of aging seems to manifest itself mainly at the level of histone modifiers and the histones themselves (in yeast there is no known role for cytosine DNA methylation in aging since its presence in the genome is generally thought to be absent [56]), where increased histone production or increased activity of certain histone modifiers, can increase lifespan [50][54][55].
–
LOSS OF PROTEOSTASIS
Protein homeostasis (proteostasis), as it pertains to aging, is generally enforced in the cell through regulation of protein production, autophagy, proteasomal degradation, and chaperone-mediated protein folding [1]. Inactivation or inefficiency of these systems results in degenerative effects, such as the formation of protein aggregations, which are generally thought of as being contributors to aging [1]. Aging-related disorders have been linked to impaired protein homeostasis, including Alzheimer’s, Parkinson’s and Huntington’s [57], and many studies have shown proteostasis to change with age [58], such as having a reduced activity in stress-induced protein chaperones [59]. Circumventing this decline by increasing chaperone protein levels has repeatedly been shown to increase longevity in worms and flies [60][61][62][63]. Recently, mice with increased autophagy levels induced by overexpression of the autophagy related protein ATG5 showed a 17% increase of lifespan [64]. Taken together, the natural decline in pathways promoting proteostasis and the influence these can have on longevity when upregulated, has earned proteostasis its rank as a hallmark of aging.
–
In yeast, to see if similar decline in chaperones occurs with aging, we can look at relevant gene ontology classes in the yeast aging proteome (Figure 2) and transcriptomes (Table 1). When observing the proteins measured for protein folding and protein targeting, we find that a slight decline also occurs with age [7] (Figure 2), and likewise decreased levels of protein folding chaperones have been described at the transcriptional level (Table 1, [65]). Contrary to this, however, a strong induction of specific chaperone proteins occurs with age, as these are part of the ‘response to stress’ gene ontology seen to be enriched with age (Table 1, [7][8]). This increase in chaperone proteins has perhaps best been illustrated by single cell studies using fluorescent reporters tracking the increased abundance levels of the protein chaperone Hsp104 [66][67]. The specific induction of chaperones that occurs with aging may reflect a targeted stress response the cell is activating, and indeed the gene ontologies for response to heat, response to osmotic stress, and response to oxidative stress (31, 30, and 44 proteins measured respectively, with partial overlap) have their proteins generally increase with age [7] (Figure 2).
–
Damage to proteins in yeast has been previously described to occur with aging in the form of increased carbonyl levels from oxidative damage [68][69]. With this, an aggregation of chaperones, namely the heat shock protein Hsp104, has been reported to occur with aging [70][71]. However, neither deletion nor up-regulation of the protein chaperone HSP104 gene has been reported to have a significant effect on lifespan [72][73], suggesting that it may indeed be the case that the cell mounts a sufficient response to sequester damaged proteins, and that the cell is not near to attaining the maximal capacity of its cytosolic protein chaperones.
–
Nonetheless, increasing protein homeostasis through an induction of the proteasome rather than chaperone activity has been demonstrated to have one of the strongest positive effects on lifespan reported in yeast [74]. This perhaps reflects an increased age-related need for proteostasis at the level of protein turnover rather than chaperone activity, and indeed, proteolysis involved in cellular protein catabolic processes slightly increases with aging [7] (Figure 2). Recently, we have shown that a loss of stoichiometry in certain protein complexes occurs with aging [7]. For example, we have noted that the nuclear pore complex (NPC) loses stoichiometry of its components with age [7], which is supported by other observations in literature that levels of the NPC components Nup116 and Nsp1 decrease relative to other components of the complex [75]. Resetting the imbalance of protein complexes that naturally occurs with aging may be one of the mechanisms by which cells slow down aging through increased proteasome activity. Further work is required to understand the exact mechanism by which increased proteasome activity increases lifespan, and the hallmark of proteostasis remains a potent modulator of yeast lifespan.
DEREGULATED NUTRIENT SENSING
Several nutrient sensing pathways have been robustly implicated in aging across multiple species [1]. These include the ‘target of rapamycin’ protein kinase TOR, which senses high amino acid concentrations (reflecting high-energy availability for the cells); the AMP activated protein kinase AMPK, which detects high AMP levels (reflecting low-energy availability for the cells); and the histone modifying sirtuins, which detect high NAD+ levels (reflecting low-energy availability for the cells) [1]. Up-regulation of the pathways sensing low-energy availability and down regulation of the pathway sensing high-energy availability results in lifespan extension [1]. Longevity benefits of some of these interventions may be mediated by the downstream forkhead box transcription factor FOXO, which activates gene transcription conferring proteotoxic stress resistance [1]. Furthermore, for multicellular organisms, the pathways of growth hormone, and the insulin and insulin-like growth factor 1 signaling, lie upstream in the nutrient sensing pathway, and both of these have been robustly shown to modulate lifespan across species [1].
–
In yeast, changes in carbon metabolism have been repeatedly described to accompany aging, and include an increase in gluconeogenesis and energy storage, and a decrease in glycolysis [26][65][76] (Table 1, Figure 2). It has been suggested that the aging yeast cell, which is continuously growing in size [77], may interpret its changing cell size to volume ratio as a cue of starvation, which in turn induces these metabolic changes [78][79]. Supporting this, when looking at aging proteome data and the 14 proteins measured in the gene ontology for response to starvation, we see a general increase in abundances to occur with aging (Figure 2). Indeed, in our recently completed analysis showing causal and responsive elements in aging, metabolic changes have been found to be downstream of earlier occurring changes [7]. Interestingly, while the cell may be interpreting cues of starvation, it remains on a trajectory of proliferation and continuous growth in cell size [80][77].
–
We previously showed that the proteome of aging yeast uncouples from the transcriptome, especially through an over-abundance of translation machinery related proteins [7]. Following this, through analysis of a high-level directional network of the aging transcriptome, we found that these translation machinery genes were a causal force, shaping the behavior of other molecular changes occurring with aging [7]. These observations suggest that the translation machinery and pathways for cellular growth are strong determinants of lifespan, and are suggestive of an aging-related hyperactivity of the nutrient sensor TOR, a regulator of translation machinery production. In line with this, deletions of genes encoding TOR1, the downstream protein kinase SCH9, and certain ribosomal proteins, increases lifespan in yeast [81][82][83]. Likewise, increasing levels of FOXO transcription factors, which is thought to confer some of the longevity benefits associated to modified nutrient sensing pathways in higher organisms [1], increases lifespan in yeast [84].
–
Nutrient sensing pathways play a role in regulating protein abundance via controlling both production and degradation of proteins. What results from this is that the hallmark of aging of deregulated nutrient sensing is closely linked to the hallmark of proteostasis. Indeed, the translation machinery, which is normally regulated by nutrient sensors, seems to be at the heart of cellular aging [7][83]. This, in addition to the fact that nutrient sensing pathways can modulate aging, and that clear signs of metabolic changes occur in aging yeast (Table 1), demonstrates that also in yeast, deregulated nutrient sensing is a hallmark of aging.
MITOCHONDRIAL DYSFUNCTION
Mitochondria have been studied in the context of aging for over half a century, having gained popularity in the field with the introduction of the free radical theory of aging [85]. The efficiency of the mitochondrial respiratory chain decreases with aging, reducing the membrane potential and ATP generation, and increasing electron leakage and production of reactive oxygen species (ROS) [1][86]. Although once thought to be purely detrimental, we now know that low levels of ROS may induce a protective homeostatic response, while only higher levels, which may increase for a variety of reasons, accelerate aging [1]. Many other changes have been proposed to contribute to aging related mitochondrial dysfunction, such as an accumulation of mutations and deletions in mtDNA, the oxidation and destabilization of mitochondrial protein complexes, a reduced biosynthesis of iron-sulfur clusters, a decline in the levels of mitochondrial biogenesis, an imbalance of fission and fusion events, and a diminishing quality control from mitophagy [87][88]. The long list of factors that are thought to contribute to increased damage and decreased turnover for mitochondria have been speculated to influence the natural aging process [1], and mitochondrial dysfunction has therefore been deemed a hallmark of aging.
–
In yeast, an early age decline in mitochondrial membrane potential has been described at ages where normally still more than 98% of the population are viable [89][90] (Figure 1). This has been accompanied by the observation that mitochondrial fragmentation occurs with aging [89][91], and decreased amounts of certain mitochondrial components are detected in aged cells [7] (Table 1, Figure 2). An aging related decline of vacuolar pH has also been associated to dysfunction of mitochondria [89]. Specifically, it was observed that vacuolar acidity drops early in aging, and is followed by the previously described structural and membrane potential changes in the mitochondria. Improving the age-related decline in vacuolar pH by overexpression of a subunit (Vma1) of the vacuolar ATPase complex that establishes acidity levels, concomitantly delays mitochondrial dysfunction and extends lifespan [89]. Though targeting the vacuole, this observation is the strongest to link interventions in mitochondrial biology in yeast with increased lifespan. Highlighting the heterogeneity of the aging phenotype though, a recent report has suggested that only a subset of the population is affected by loss of mitochondrial membrane potential in this way [9]. A second observation linking change in mitochondria to longevity is the occurrence of petites, cells which are defective in respiration [92]. The transformation of a yeast cell into a petite can result in an increased lifespan for the cell under certain, but not all, conditions [93]. Interestingly, an increase in the rate of formation of petite daughter cells may be occurring during aging [92], which could coincide with observations that mtDNA translocate to the nuclear genome in mother cells of advanced age [30].
–
Mitochondrial malfunction has the potential to cause detrimental effects in the yeast cell. Increased ROS levels have been observed to occur with aging [66], as have levels of ROS specifically localized to the mitochondria [94]. This may explain why oxidative damage to proteins increases markedly in aging yeast [69][71], and why a strong response to oxidative damage and oxidative stress is induced [68][71]. Overall, clear signs are present that mitochondrial dysfunction is a hallmark of yeast aging, from altered mitochondrial morphology and membrane potential, to the production of ROS and the increasing damage it causes to proteins [69][71].
DISCUSSION
We have conducted a tour of the literature to shed light on the aging process in yeast, and have found clear signs that the majority of hallmarks of aging present in vertebrates also manifest themselves in replicatively aging yeast. Additionally, we note that other phenotypes of aging exist in yeast that may not readily classify under a specific hallmark. For example, we see that proteins in the gene ontology for protein phosphorylation and protein dephosphorylation appear to undergo opposing abundance changes with age [7] (Figure 2), suggesting an overall increase in phosphorylation state. The pathways of endocytosis, membrane fusion, and organelle fission, fusion and inheritance are all seen to undergo diverse changes with age at the protein level [7] (Figure 2), pointing towards a potentially important role in aging. Also, the yeast vacuole seems greatly affected as proteins involved in its organization increase with age [7] (Figure 2), certain vacuolar proteins preferentially accumulate in mother cells [95], and the vacuole enlarges with signs of invagination [80][96]. Other changes occurring in aging yeast include a reduction in sporulation efficiency [97], an increasingly random budding pattern [98], an increase in sterility [14], an enlarging nucleolus and increased G2/M DNA content [55][70], and either increased or decreased sensitivity to drugs, stresses, and mutagens [99][100][101] (Figure 1). Studying why and how these aging related phenotypes happen in yeast may provide interesting avenues of future research in higher organisms. Clearly, there is still more to be gained from yeast for understanding human aging.
–
The hallmarks of aging as presented in [1] were chosen partially based on their ability to confer extended longevity with interventions. Likewise, in our tour of literature, we have found this to be the case for the hallmarks of human aging that are applicable to yeast. For each hallmark, specific interventions can extend lifespan of yeast. For genomic instability, deleting FOB1 reduces ERC formation and extends longevity [33]. For epigenetic modifications, increasing levels of Sir2 [50], histones H3 and H4 [55], or deletion of genes encoding the SAGA components SGF73, SGF11, and UBP8 [54], results in a modified epigenome and extended longevity. For the hallmark of proteostasis, deletion of UBR2 was shown to increase lifespan by increasing proteasome activity [74]. For deregulated nutrient sensing deletions of TOR1 or SCH9 increase lifespan [82][83] as does dietary restriction (DR) mimetics such as the HXK2 deletion strain [102], and DR itself under certain, but not all, conditions [102][103][104]. Likewise, FOXO, thought to be downstream in many nutrient-sensing pathways that increase lifespan when modulated, increases lifespan in yeast [84]. Finally, for the hallmark of mitochondrial dysfunction, improving mitochrondrial function by overexpression of the vacuolar ATPase protein Vma1 results in lifespan extension [89].
–
Interestingly, as depicted in Figure 1, many of the phenotypes of aging manifest themselves early in the yeast’s lifespan, while still 90% of the population remains viable. The presence of many early-age phenotypes in literature is partially due to the fact that enriching yeast cultures for cells of advanced age is a difficult process, and it is therefore easier to characterize younger ‘aged’ cells [2]. Despite this, studies that have explored into deeper ages in the population’s viability have nonetheless found relatively similar changes at the process level to occur in middle and old ages (Table 1). Indeed, we previously found that most changes occur early and are progressive over time [7]. Since most of the studies characterizing aging cells were done at the population level, interesting questions that arise are whether the occurrence of these phenotypes are delayed in naturally longer-lived cells within a population, if these longer lived cells may be more resistant to certain changes, or if they may have different pathologies and causes of mortality than naturally shorter lived cells. Recently, these questions have drawn the attention of researchers and are beginning to be answered [9][80][66][67][105].
–
As discussed in a dedicated review [106] the change in one phenotype impacts many others, leading to either the sequential collapse of different functions or the triggering of compensating responses making up for the decline of one subsystem. To begin to map these connections on a system wide level, we predicted based on network analysis that the changes in the genes related to protein biogenesis are causal to diverse other age related changes, putting the hallmark of protein homeostasis at the center of attention. Indeed, system-level phenotypes of aging in yeast [7], have been shown to be conserved in humans and rhesus macaques (brain prefrontal cortex [107]) and rat (brain, liver [108]) and include a progressive uncoupling of the proteome from the transcriptionally encoded message, and a change in stoichiometry of the components of certain protein complexes.
–
In studies more dedicated to understanding individual molecular mechanisms and pathways of aging, elegant examples of the interconnected nature of the aging process can be found. For example, one sequence of events involving the plasma membrane, vacuole, and mitochondria and linking to genome stability bridges several hallmarks of aging, as follows. The sequence starts with the activity of the plasma membrane ATPase, Pma1, which already early in life causes the cytosolic pH of yeast to increase [109]. This activity antagonizes vacuole acidity by reducing cytosolic protons, which results in an increased vacuolar pH. This, in turn, compromises storage of neutral amino acids [89]. An excess of these in the cytosol may overburden the mitochondrial dependent catabolism of amino acids, which could be a cause of the loss in mitochondrial membrane potential [89]. A loss in mitochondrial membrane potential culminates in a reduced synthesis of iron-sulfur clusters [92], and, as these are required as cofactors in many DNA repair enzymes, the entire cascade of events thus links to genomic stability [27].
–
Interestingly, overexpression of only one of the component of the vacuolar proton ATPase, VMA1, results in lifespan extension and rescue of some of the above mentioned phenotypes [89]. The vacuolar proton ATPase is one of the complexes in a yeast cell that shows a clear loss of its stoichiometry in replicative aging, and indeed the protein abundance of VMA1 decreases relative to most other components [7]. We speculate that the rescue of the aging phenotypes from overexpression of VMA1 is thus related to the general problem of maintaining protein complex stoichiometry in aging.
–
In some cases feed forward loops may also present themselves, driving cell death. E.g. the nuclear pore complex shows a strong loss of the stoichiometry of its protein components [7] which may be a cause of altered transport function, impacting ribosome biogenesis, mRNA export, and import of transcriptional regulators and tRNA, all feeding back into the potential causes of the NPC sub-stoichiometry. Finding how the different hallmarks are dependent on each other remains an important task for the future to understand the aging process.
–
Overall, we find evidence that the hallmarks involved in vertebrate aging are implicated in the replicative aging of yeast, indicating that aging is affected by dysfunction of shared processes. Nutrient sensing and mitochondria are vital for activity of the cell, while proteostasis is integral to the maintenance of the cellular system. Meanwhile the very identity of the cell, the genome and epigenome, are essential to carry the message for what processes and functions should occur. These processes are universally necessary to support cell growth, viability and division in all eukaryotic life, from yeast to vertebrates, and it follows that their dysfunction should be integral to disease and aging.
References
- C. López-Otín, M.A. Blasco, L. Partridge, M. Serrano, and G. Kroemer, "The Hallmarks of Aging", Cell, vol. 153, pp. 1194-1217, 2013. http://dx.doi.org/10.1016/j.cell.2013.05.039
- A. Denoth Lippuner, T. Julou, and Y. Barral, "Budding yeast as a model organism to study the effects of age", FEMS Microbiology Reviews, vol. 38, pp. 300-325, 2014. http://dx.doi.org/10.1111/1574-6976.12060
- B.M. Wasko, and M. Kaeberlein, "Yeast replicative aging: a paradigm for defining conserved longevity interventions", FEMS Yeast Research, vol. 14, pp. 148-159, 2013. http://dx.doi.org/10.1111/1567-1364.12104
- K. Steinkraus, M. Kaeberlein, and B. Kennedy, "Replicative Aging in Yeast: The Means to the End", Annual Review of Cell and Developmental Biology, vol. 24, pp. 29-54, 2008. http://dx.doi.org/10.1146/annurev.cellbio.23.090506.123509
- P. Fabrizio, and V.D. Longo, "The Chronological Life Span of Saccharomyces cerevisiae", Methods in Molecular Biology, pp. 89-95, 2007. http://dx.doi.org/10.1007/978-1-59745-361-5_8
- V. Longo, G. Shadel, M. Kaeberlein, and B. Kennedy, "Replicative and Chronological Aging in Saccharomyces cerevisiae", Cell Metabolism, vol. 16, pp. 18-31, 2012. http://dx.doi.org/10.1016/j.cmet.2012.06.002
- G.E. Janssens, A.C. Meinema, J. González, J.C. Wolters, A. Schmidt, V. Guryev, R. Bischoff, E.C. Wit, L.M. Veenhoff, and M. Heinemann, "Protein biogenesis machinery is a driver of replicative aging in yeast", eLife, vol. 4, 2015. http://dx.doi.org/10.7554/eLife.08527
- P. LAUN, L. RAMACHANDRAN, S. JAROLIM, E. HERKER, P. LIANG, J. WANG, M. WEINBERGER, D. BURHANS, B. SUTER, and F. MADEO, "A comparison of the aging and apoptotic transcriptome of", FEMS Yeast Research, vol. 5, pp. 1261-1272, 2005. http://dx.doi.org/10.1016/j.femsyr.2005.07.006
- S. Fehrmann, C. Paoletti, Y. Goulev, A. Ungureanu, H. Aguilaniu, and G. Charvin, "Aging Yeast Cells Undergo a Sharp Entry into Senescence Unrelated to the Loss of Mitochondrial Membrane Potential", Cell Reports, vol. 5, pp. 1589-1599, 2013. http://dx.doi.org/10.1016/j.celrep.2013.11.013
- I.M. Conboy, and T.A. Rando, "Heterochronic parabiosis for the study of the effects of aging on stem cells and their niches", Cell Cycle, vol. 11, pp. 2260-2267, 2012. http://dx.doi.org/10.4161/cc.20437
- N.R. Austriaco, and L.P. Guarente, "Changes of telomere length cause reciprocal changes in the lifespan of mother cells in Saccharomyces cerevisiae", Proceedings of the National Academy of Sciences, vol. 94, pp. 9768-9772, 1997. http://dx.doi.org/10.1073/pnas.94.18.9768
- S. Kim, B. Villeponteau, and S. Jazwinski, "Effect of Replicative Age on Transcriptional Silencing Near Telomeres inSaccharomyces cerevisiae", Biochemical and Biophysical Research Communications, vol. 219, pp. 370-376, 1996. http://dx.doi.org/10.1006/bbrc.1996.0240
- N.P. D'Mello, and S.M. Jazwinski, "Telomere length constancy during aging of Saccharomyces cerevisiae.", Journal of bacteriology, 1991. http://www.ncbi.nlm.nih.gov/pubmed/1938877
- I. M�ller, "Parental age and the life-span of zygotes of Saccharomyces cerevisiae", Antonie van Leeuwenhoek, vol. 51, pp. 1-10, 1985. http://dx.doi.org/10.1007/BF00444223
- T. Smeal, J. Claus, B. Kennedy, F. Cole, and L. Guarente, "Loss of Transcriptional Silencing Causes Sterility in Old Mother Cells of S. cerevisiae", Cell, vol. 84, pp. 633-642, 1996. http://dx.doi.org/10.1016/S0092-8674(00)81038-7
- S. Mei, and C. Brenner, "Calorie Restriction-Mediated Replicative Lifespan Extension in Yeast Is Non-Cell Autonomous", PLOS Biology, vol. 13, pp. e1002048, 2015. http://dx.doi.org/10.1371/journal.pbio.1002048
- J. Vijg, and Y. Suh, "Genome Instability and Aging", Annual Review of Physiology, vol. 75, pp. 645-668, 2013. http://dx.doi.org/10.1146/annurev-physiol-030212-183715
- G. Failla, "THE AGING PROCESS AND CANCEROGENESIS", Annals of the New York Academy of Sciences, vol. 71, pp. 1124-1140, 1958. http://dx.doi.org/10.1111/j.1749-6632.1958.tb46828.x
- L. Szilard, "ON THE NATURE OF THE AGING PROCESS", Proceedings of the National Academy of Sciences, vol. 45, pp. 30-45, 1959. http://dx.doi.org/10.1073/pnas.45.1.30
- J.H. Hoeijmakers, "DNA Damage, Aging, and Cancer", New England Journal of Medicine, vol. 361, pp. 1475-1485, 2009. http://dx.doi.org/10.1056/NEJMra0804615
- A.A. Moskalev, M.V. Shaposhnikov, E.N. Plyusnina, A. Zhavoronkov, A. Budovsky, H. Yanai, and V.E. Fraifeld, "The role of DNA damage and repair in aging through the prism of Koch-like criteria", Ageing Research Reviews, vol. 12, pp. 661-684, 2013. http://dx.doi.org/10.1016/j.arr.2012.02.001
- C.R. Burtner, and B.K. Kennedy, "Progeria syndromes and ageing: what is the connection?", Nature Reviews Molecular Cell Biology, vol. 11, pp. 567-578, 2010. http://dx.doi.org/10.1038/nrm2944
- R.D. Kolodner, C.D. Putnam, and K. Myung, "Maintenance of Genome Stability in Saccharomyces cerevisiae", Science, vol. 297, pp. 552-557, 2002. http://dx.doi.org/10.1126/science.1075277
- ""http://www.ncbi.nlm.nih.gov/pubmed/112907010
- L.L.M. Hoopes, M. Budd, W. Choe, T. Weitao, and J.L. Campbell, "Mutations in DNA Replication Genes Reduce Yeast Life Span", Molecular and Cellular Biology, vol. 22, pp. 4136-4146, 2002. http://dx.doi.org/10.1128/MCB.22.12.4136-4146.2002
- I. Lesur, and J.L. Campbell, "The Transcriptome of Prematurely Aging Yeast Cells Is Similar to That of Telomerase-deficient Cells", Molecular Biology of the Cell, vol. 15, pp. 1297-1312, 2004. http://dx.doi.org/10.1091/mbc.E03-10-0742
- M.A. McMurray, and D.E. Gottschling, "An Age-Induced Switch to a Hyper-Recombinational State", Science, vol. 301, pp. 1908-1911, 2003. http://dx.doi.org/10.1126/science.1087706
- S. Gravel, and S.P. Jackson, "Increased Genome Instability in Aging Yeast", Cell, vol. 115, pp. 1-2, 2003. http://dx.doi.org/10.1016/s0092-8674(03)00766-9
- B.K. Kennedy, N.R. Austriaco, and L. Guarente, "Daughter cells of Saccharomyces cerevisiae from old mothers display a reduced life span.", The Journal of cell biology, vol. 127, pp. 1985-1993, 1994. http://dx.doi.org/10.1083/jcb.127.6.1985
- Z. Hu, K. Chen, Z. Xia, M. Chavez, S. Pal, J. Seol, C. Chen, W. Li, and J.K. Tyler, "Nucleosome loss leads to global transcriptional up-regulation and genomic instability during yeast aging", Genes & Development, vol. 28, pp. 396-408, 2014. http://dx.doi.org/10.1101/gad.233221.113
- A. Kaya, A.V. Lobanov, and V.N. Gladyshev, "Evidence that mutation accumulation does not cause aging in Saccharomyces cerevisiae", Aging Cell, vol. 14, pp. 366-371, 2015. http://dx.doi.org/10.1111/acel.12290
- D.A. Sinclair, and L. Guarente, "Extrachromosomal rDNA Circles— A Cause of Aging in Yeast", Cell, vol. 91, pp. 1033-1042, 1997. http://dx.doi.org/10.1016/S0092-8674(00)80493-6
- P. Defossez, R. Prusty, M. Kaeberlein, S. Lin, P. Ferrigno, P.A. Silver, R.L. Keil, and L. Guarente, "Elimination of Replication Block Protein Fob1 Extends the Life Span of Yeast Mother Cells", Molecular Cell, vol. 3, pp. 447-455, 1999. http://dx.doi.org/10.1016/S1097-2765(00)80472-4
- A.A. Falcón, and J.P. Aris, "Plasmid Accumulation Reduces Life Span in Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 278, pp. 41607-41617, 2003. http://dx.doi.org/10.1074/jbc.M307025200
- T. Kobayashi, "A new role of the rDNA and nucleolus in the nucleus—rDNA instability maintains genome integrity", BioEssays, vol. 30, pp. 267-272, 2008. http://dx.doi.org/10.1002/bies.20723
- A.R. Ganley, S. Ide, K. Saka, and T. Kobayashi, "The Effect of Replication Initiation on Gene Amplification in the rDNA and Its Relationship to Aging", Molecular Cell, vol. 35, pp. 683-693, 2009. http://dx.doi.org/10.1016/j.molcel.2009.07.012
- K. Saka, S. Ide, A. Ganley, and T. Kobayashi, "Cellular Senescence in Yeast Is Regulated by rDNA Noncoding Transcription", Current Biology, vol. 23, pp. 1794-1798, 2013. http://dx.doi.org/10.1016/j.cub.2013.07.048
- E.X. Kwan, E.J. Foss, S. Tsuchiyama, G.M. Alvino, L. Kruglyak, M. Kaeberlein, M.K. Raghuraman, B.J. Brewer, B.K. Kennedy, and A. Bedalov, "A Natural Polymorphism in rDNA Replication Origins Links Origin Activation with Calorie Restriction and Lifespan", PLoS Genetics, vol. 9, pp. e1003329, 2013. http://dx.doi.org/10.1371/journal.pgen.1003329
- A.R.D. Ganley, and T. Kobayashi, "Ribosomal DNA and cellular senescence: new evidence supporting the connection between rDNA and aging", FEMS Yeast Research, vol. 14, pp. 49-59, 2014. http://dx.doi.org/10.1111/1567-1364.12133
- R. Sweeney, and V.A. Zakian, "Extrachromosomal elements cause a reduced division potential in nib 1 strains of Saccharomyces cerevisiae.", Genetics, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2668119
- B. Sarg, E. Koutzamani, W. Helliger, I. Rundquist, and H.H. Lindner, "Postsynthetic Trimethylation of Histone H4 at Lysine 20 in Mammalian Tissues Is Associated with Aging", Journal of Biological Chemistry, vol. 277, pp. 39195-39201, 2002. http://dx.doi.org/10.1074/jbc.M205166200
- I. Cheung, H.P. Shulha, Y. Jiang, A. Matevossian, J. Wang, Z. Weng, and S. Akbarian, "Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex", Proceedings of the National Academy of Sciences, vol. 107, pp. 8824-8829, 2010. http://dx.doi.org/10.1073/pnas.1001702107
- M.F. Fraga, and M. Esteller, "Epigenetics and aging: the targets and the marks", Trends in Genetics, vol. 23, pp. 413-418, 2007. http://dx.doi.org/10.1016/j.tig.2007.05.008
- S. Han, and A. Brunet, "Histone methylation makes its mark on longevity", Trends in Cell Biology, vol. 22, pp. 42-49, 2012. http://dx.doi.org/10.1016/j.tcb.2011.11.001
- E.L. Greer, T.J. Maures, A.G. Hauswirth, E.M. Green, D.S. Leeman, G.S. Maro, S. Han, M.R. Banko, O. Gozani, and A. Brunet, "Members of the H3K4 trimethylation complex regulate lifespan in a germline-dependent manner in C. elegans", Nature, vol. 466, pp. 383-387, 2010. http://dx.doi.org/10.1038/nature09195
- A.P. Siebold, R. Banerjee, F. Tie, D.L. Kiss, J. Moskowitz, and P.J. Harte, "Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance", Proceedings of the National Academy of Sciences, vol. 107, pp. 169-174, 2009. http://dx.doi.org/10.1073/pnas.0907739107
- C. Jin, J. Li, C. Green, X. Yu, X. Tang, D. Han, B. Xian, D. Wang, X. Huang, X. Cao, Z. Yan, L. Hou, J. Liu, N. Shukeir, P. Khaitovich, C. Chen, H. Zhang, T. Jenuwein, and J. Han, "Histone Demethylase UTX-1 Regulates C. elegans Life Span by Targeting the Insulin/IGF-1 Signaling Pathway", Cell Metabolism, vol. 14, pp. 161-172, 2011. http://dx.doi.org/10.1016/j.cmet.2011.07.001
- R.E. Marioni, S. Shah, A.F. McRae, B.H. Chen, E. Colicino, S.E. Harris, J. Gibson, A.K. Henders, P. Redmond, S.R. Cox, A. Pattie, J. Corley, L. Murphy, N.G. Martin, G.W. Montgomery, A.P. Feinberg, M.D. Fallin, M.L. Multhaup, A.E. Jaffe, R. Joehanes, J. Schwartz, A.C. Just, K.L. Lunetta, J.M. Murabito, J.M. Starr, S. Horvath, A.A. Baccarelli, D. Levy, P.M. Visscher, N.R. Wray, and I.J. Deary, "DNA methylation age of blood predicts all-cause mortality in later life", Genome Biology, vol. 16, 2015. http://dx.doi.org/10.1186/s13059-015-0584-6
-
M.J. Jones, S.J. Goodman, and M.S. Kobor, "
DNA methylation and healthy human aging", Aging Cell, vol. 14, pp. 924-932, 2015. http://dx.doi.org/10.1111/acel.12349 - M. Kaeberlein, M. McVey, and L. Guarente, "The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms", Genes & Development, vol. 13, pp. 2570-2580, 1999. http://dx.doi.org/10.1101/gad.13.19.2570
- L. Guarente, "Sirtuins, Aging, and Metabolism", Cold Spring Harbor Symposia on Quantitative Biology, vol. 76, pp. 81-90, 2011. http://dx.doi.org/10.1101/sqb.2011.76.010629
- W. Dang, K.K. Steffen, R. Perry, J.A. Dorsey, F.B. Johnson, A. Shilatifard, M. Kaeberlein, B.K. Kennedy, and S.L. Berger, "Histone H4 lysine 16 acetylation regulates cellular lifespan", Nature, vol. 459, pp. 802-807, 2009. http://dx.doi.org/10.1038/nature08085
- N. Suka, K. Luo, and M. Grunstein, "Sir2p and Sas2p opposingly regulate acetylation of yeast histone H4 lysine16 and spreading of heterochromatin", Nature Genetics, vol. 32, pp. 378-383, 2002. http://dx.doi.org/10.1038/ng1017
- M. McCormick, A. Mason, S. Guyenet, W. Dang, R. Garza, M. Ting, R. Moller, S. Berger, M. Kaeberlein, L. Pillus, A. La Spada, and B. Kennedy, "The SAGA Histone Deubiquitinase Module Controls Yeast Replicative Lifespan via Sir2 Interaction", Cell Reports, vol. 8, pp. 477-486, 2014. http://dx.doi.org/10.1016/j.celrep.2014.06.037
- J. Feser, D. Truong, C. Das, J.J. Carson, J. Kieft, T. Harkness, and J.K. Tyler, "Elevated Histone Expression Promotes Life Span Extension", Molecular Cell, vol. 39, pp. 724-735, 2010. http://dx.doi.org/10.1016/j.molcel.2010.08.015
- F. Capuano, M. Mülleder, R. Kok, H.J. Blom, and M. Ralser, "Cytosine DNA Methylation Is Found in Drosophila melanogaster but Absent in Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Other Yeast Species", Analytical Chemistry, vol. 86, pp. 3697-3702, 2014. http://dx.doi.org/10.1021/ac500447w
- I. Saez, and D. Vilchez, "The Mechanistic Links Between Proteasome Activity, Aging and Agerelated Diseases", Current Genomics, vol. 15, pp. 38-51, 2014. http://dx.doi.org/10.2174/138920291501140306113344
- H. Koga, S. Kaushik, and A.M. Cuervo, "Protein homeostasis and aging: The importance of exquisite quality control", Ageing Research Reviews, vol. 10, pp. 205-215, 2011. http://dx.doi.org/10.1016/j.arr.2010.02.001
- S.K. Calderwood, A. Murshid, and T. Prince, "The Shock of Aging: Molecular Chaperones and the Heat Shock Response in Longevity and Aging – A Mini-Review", Gerontology, vol. 55, pp. 550-558, 2009. http://dx.doi.org/10.1159/000225957
- G. Morrow, M. Samson, S. Michaud, and R. M. Tanguay, "Overexpression of the small mitochondrial Hsp22 extendsDrosophilalife span and increases resistance to oxidative stress", The FASEB Journal, vol. 18, pp. 598-599, 2004. http://dx.doi.org/10.1096/fj.03-0860fje
- G.A. Walker, and G.J. Lithgow, "Lifespan extension in C. elegans by a molecular chaperone dependent upon insulin‐like signals", Aging Cell, vol. 2, pp. 131-139, 2003. http://dx.doi.org/10.1046/j.1474-9728.2003.00045.x
- W. Chiang, T. Ching, H. Lee, C. Mousigian, and A. Hsu, "HSF-1 Regulators DDL-1/2 Link Insulin-like Signaling to Heat-Shock Responses and Modulation of Longevity", Cell, vol. 148, pp. 322-334, 2012. http://dx.doi.org/10.1016/j.cell.2011.12.019
- A. Hsu, C.T. Murphy, and C. Kenyon, "Regulation of Aging and Age-Related Disease by DAF-16 and Heat-Shock Factor", Science, vol. 300, pp. 1142-1145, 2003. http://dx.doi.org/10.1126/science.1083701
- J. Pyo, S. Yoo, H. Ahn, J. Nah, S. Hong, T. Kam, S. Jung, and Y. Jung, "Overexpression of Atg5 in mice activates autophagy and extends lifespan", Nature Communications, vol. 4, 2013. http://dx.doi.org/10.1038/ncomms3300
- A. Koc, A.P. Gasch, J.C. Rutherford, H. Kim, and V.N. Gladyshev, "Methionine sulfoxide reductase regulation of yeast lifespan reveals reactive oxygen species-dependent and -independent components of aging", Proceedings of the National Academy of Sciences, vol. 101, pp. 7999-8004, 2004. http://dx.doi.org/10.1073/pnas.0307929101
- Z. Xie, Y. Zhang, K. Zou, O. Brandman, C. Luo, Q. Ouyang, and H. Li, "Molecular phenotyping of aging in single yeast cells using a novel microfluidic device", Aging Cell, vol. 11, pp. 599-606, 2012. http://dx.doi.org/10.1111/j.1474-9726.2012.00821.x
- M.M. Crane, I.B.N. Clark, E. Bakker, S. Smith, and P.S. Swain, "A Microfluidic System for Studying Ageing and Dynamic Single-Cell Responses in Budding Yeast", PLoS ONE, vol. 9, pp. e100042, 2014. http://dx.doi.org/10.1371/journal.pone.0100042
- G. Reverter-Branchat, E. Cabiscol, J. Tamarit, and J. Ros, "Oxidative Damage to Specific Proteins in Replicative and Chronological-aged Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 279, pp. 31983-31989, 2004. http://dx.doi.org/10.1074/jbc.M404849200
- H. Aguilaniu, L. Gustafsson, M. Rigoulet, and T. Nyström, "Asymmetric Inheritance of Oxidatively Damaged Proteins During Cytokinesis", Science, vol. 299, pp. 1751-1753, 2003. http://dx.doi.org/10.1126/science.1080418
- E. Ünal, B. Kinde, and A. Amon, "Gametogenesis Eliminates Age-Induced Cellular Damage and Resets Life Span in Yeast", Science, vol. 332, pp. 1554-1557, 2011. http://dx.doi.org/10.1126/science.1204349
- N. Erjavec, L. Larsson, J. Grantham, and T. Nyström, "Accelerated aging and failure to segregate damaged proteins in Sir2 mutants can be suppressed by overproducing the protein aggregation-remodeling factor Hsp104p", Genes & Development, vol. 21, pp. 2410-2421, 2007. http://dx.doi.org/10.1101/gad.439307
- M. Kaeberlein, K.T. Kirkland, S. Fields, and B.K. Kennedy, "Genes determining yeast replicative life span in a long-lived genetic background", Mechanisms of Ageing and Development, vol. 126, pp. 491-504, 2005. http://dx.doi.org/10.1016/j.mad.2004.10.007
- V. Andersson, S. Hanzén, B. Liu, M. Molin, and T. Nyström, "Enhancing protein disaggregation restores proteasome activity in aged cells.", Aging, 2013. http://www.ncbi.nlm.nih.gov/pubmed/24243762
- U. Kruegel, B. Robison, T. Dange, G. Kahlert, J.R. Delaney, S. Kotireddy, M. Tsuchiya, S. Tsuchiyama, C.J. Murakami, J. Schleit, G. Sutphin, D. Carr, K. Tar, G. Dittmar, M. Kaeberlein, B.K. Kennedy, and M. Schmidt, "Elevated Proteasome Capacity Extends Replicative Lifespan in Saccharomyces cerevisiae", PLoS Genetics, vol. 7, pp. e1002253, 2011. http://dx.doi.org/10.1371/journal.pgen.1002253
- C.L. Lord, B.L. Timney, M.P. Rout, and S.R. Wente, "Altering nuclear pore complex function impacts longevity and mitochondrial function in S. cerevisiae", Journal of Cell Biology, vol. 208, pp. 729-744, 2015. http://dx.doi.org/10.1083/jcb.201412024
- S.S. Lin, J.K. Manchester, and J.I. Gordon, "Enhanced Gluconeogenesis and Increased Energy Storage as Hallmarks of Aging in Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 276, pp. 36000-36007, 2001. http://dx.doi.org/10.1074/jbc.M103509200
- N.K. Egilmez, J.B. Chen, and S.M. Jazwinski, "Preparation and Partial Characterization of Old Yeast Cells", Journal of Gerontology, vol. 45, pp. B9-B17, 1990. http://dx.doi.org/10.1093/geronj/45.1.B9
- G. Yiu, A. McCord, A. Wise, R. Jindal, J. Hardee, A. Kuo, M.Y. Shimogawa, L. Cahoon, M. Wu, J. Kloke, J. Hardin, and L.L. Mays Hoopes, "Pathways Change in Expression During Replicative Aging in Saccharomyces cerevisiae", The Journals of Gerontology Series A: Biological Sciences and Medical Sciences, vol. 63, pp. 21-34, 2008. http://dx.doi.org/10.1093/gerona/63.1.21
- Y. Kamei, Y. Tamada, Y. Nakayama, E. Fukusaki, and Y. Mukai, "Changes in Transcription and Metabolism During the Early Stage of Replicative Cellular Senescence in Budding Yeast", Journal of Biological Chemistry, vol. 289, pp. 32081-32093, 2014. http://dx.doi.org/10.1074/jbc.M114.600528
- S.S. Lee, I.A. Vizcarra, D.H.E.W. Huberts, L.P. Lee, and M. Heinemann, "Whole lifespan microscopic observation of budding yeast aging through a microfluidic dissection platform", Proceedings of the National Academy of Sciences, vol. 109, pp. 4916-4920, 2012. http://dx.doi.org/10.1073/pnas.1113505109
- K.K. Steffen, V.L. MacKay, E.O. Kerr, M. Tsuchiya, D. Hu, L.A. Fox, N. Dang, E.D. Johnston, J.A. Oakes, B.N. Tchao, D.N. Pak, S. Fields, B.K. Kennedy, and M. Kaeberlein, "Yeast Life Span Extension by Depletion of 60S Ribosomal Subunits Is Mediated by Gcn4", Cell, vol. 133, pp. 292-302, 2008. http://dx.doi.org/10.1016/j.cell.2008.02.037
- M. Kaeberlein, R.W. Powers, K.K. Steffen, E.A. Westman, D. Hu, N. Dang, E.O. Kerr, K.T. Kirkland, S. Fields, and B.K. Kennedy, "Regulation of Yeast Replicative Life Span by TOR and Sch9 in Response to Nutrients", Science, vol. 310, pp. 1193-1196, 2005. http://dx.doi.org/10.1126/science.1115535
- S.C. Johnson, P.S. Rabinovitch, and M. Kaeberlein, "mTOR is a key modulator of ageing and age-related disease", Nature, vol. 493, pp. 338-345, 2013. http://dx.doi.org/10.1038/nature11861
- S.D.L. Postnikoff, M.E. Malo, B. Wong, and T.A.A. Harkness, "The Yeast Forkhead Transcription Factors Fkh1 and Fkh2 Regulate Lifespan and Stress Response Together with the Anaphase-Promoting Complex", PLoS Genetics, vol. 8, pp. e1002583, 2012. http://dx.doi.org/10.1371/journal.pgen.1002583
- D. Harman, "Aging: A Theory Based on Free Radical and Radiation Chemistry", Journal of Gerontology, vol. 11, pp. 298-300, 1956. http://dx.doi.org/10.1093/geronj/11.3.298
- D.R. Green, L. Galluzzi, and G. Kroemer, "Mitochondria and the Autophagy–Inflammation–Cell Death Axis in Organismal Aging", Science, vol. 333, pp. 1109-1112, 2011. http://dx.doi.org/10.1126/science.1201940
- K. Wang, and D.J. Klionsky, "Mitochondria removal by autophagy", Autophagy, vol. 7, pp. 297-300, 2011. http://dx.doi.org/10.4161/auto.7.3.14502
- M. Breitenbach, M. Rinnerthaler, J. Hartl, A. Stincone, J. Vowinckel, H. Breitenbach-Koller, and M. Ralser, "Mitochondria in ageing: there is metabolism beyond the ROS", FEMS Yeast Research, vol. 14, pp. 198-212, 2014. http://dx.doi.org/10.1111/1567-1364.12134
- A.L. Hughes, and D.E. Gottschling, "An early age increase in vacuolar pH limits mitochondrial function and lifespan in yeast", Nature, vol. 492, pp. 261-265, 2012. http://dx.doi.org/10.1038/nature11654
- J.R. McFaline‐Figueroa, J. Vevea, T.C. Swayne, C. Zhou, C. Liu, G. Leung, I.R. Boldogh, and L.A. Pon, "Mitochondrial quality control during inheritance is associated with lifespan and mother–daughter age asymmetry in budding yeast", Aging Cell, vol. 10, pp. 885-895, 2011. http://dx.doi.org/10.1111/j.1474-9726.2011.00731.x
- D.H.E.W. Huberts, G.E. Janssens, S.S. Lee, I.A. Vizcarra, and M. Heinemann, "Continuous High-resolution Microscopic Observation of Replicative Aging in Budding Yeast", Journal of Visualized Experiments, 2013. http://dx.doi.org/10.3791/50143
- J.R. Veatch, M.A. McMurray, Z.W. Nelson, and D.E. Gottschling, "Mitochondrial Dysfunction Leads to Nuclear Genome Instability via an Iron-Sulfur Cluster Defect", Cell, vol. 137, pp. 1247-1258, 2009. http://dx.doi.org/10.1016/j.cell.2009.04.014
- P.A. Kirchman, S. Kim, C.Y. Lai, and S.M. Jazwinski, "Interorganelle signaling is a determinant of longevity in Saccharomyces cerevisiae.", Genetics, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10224252
- P. Laun, A. Pichova, F. Madeo, J. Fuchs, A. Ellinger, S. Kohlwein, I. Dawes, K. Fröhlich, and M. Breitenbach, "Aged mother cells of Saccharomyces cerevisiae show markers of oxidative stress and apoptosis", Molecular Microbiology, vol. 39, pp. 1166-1173, 2001. http://dx.doi.org/10.1111/j.1365-2958.2001.02317.x
- J. Yang, M.A. McCormick, J. Zheng, Z. Xie, M. Tsuchiya, S. Tsuchiyama, H. El-Samad, Q. Ouyang, M. Kaeberlein, B.K. Kennedy, and H. Li, "Systematic analysis of asymmetric partitioning of yeast proteome between mother and daughter cells reveals “aging factors” and mechanism of lifespan asymmetry", Proceedings of the National Academy of Sciences, vol. 112, pp. 11977-11982, 2015. http://dx.doi.org/10.1073/pnas.1506054112
- F. Tang, J.W. Watkins, M. Bermudez, R. Gray, A. Gaban, K. Portie, S. Grace, M. Kleve, and G. Craciun, "A lifespan-extending form of autophagy employs the vacuole-vacuole fusion machinery", Autophagy, vol. 4, pp. 874-886, 2008. http://dx.doi.org/10.4161/auto.6556
- M. Boselli, J. Rock, E. Ünal, S.S. Levine, and A. Amon, "Effects of Age on Meiosis in Budding Yeast", Developmental Cell, vol. 16, pp. 844-855, 2009. http://dx.doi.org/10.1016/j.devcel.2009.05.013
- S. Jazwinski, S. Kim, C. Lai, and A. Benguria, "Epigenetic stratification: the role of individual change in the biological aging process", Experimental Gerontology, vol. 33, pp. 571-580, 1998. http://dx.doi.org/10.1016/S0531-5565(98)00029-1
- A. Eldakak, G. Rancati, B. Rubinstein, P. Paul, V. Conaway, and R. Li, "Asymmetrically inherited multidrug resistance transporters are recessive determinants in cellular replicative ageing", Nature Cell Biology, vol. 12, pp. 799-805, 2010. http://dx.doi.org/10.1038/ncb2085
- S.F. Levy, N. Ziv, and M.L. Siegal, "Bet Hedging in Yeast by Heterogeneous, Age-Correlated Expression of a Stress Protectant", PLoS Biology, vol. 10, pp. e1001325, 2012. http://dx.doi.org/10.1371/journal.pbio.1001325
- S.P. Kale, and S.M. Jazwinski, "Differential response to UV stress and DNA damage during the yeast replicative life span", Developmental Genetics, vol. 18, pp. 154-160, 1996. http://dx.doi.org/10.1002/(sici)1520-6408(1996)18:2<154::aid-dvg8>3.0.co;2-8
- S. Lin, P. Defossez, and L. Guarente, "Requirement of NAD and SIR2 for Life-Span Extension by Calorie Restriction in Saccharomyces cerevisiae", Science, vol. 289, pp. 2126-2128, 2000. http://dx.doi.org/10.1126/science.289.5487.2126
- D.H.E.W. Huberts, J. González, S.S. Lee, A. Litsios, G. Hubmann, E.C. Wit, and M. Heinemann, "Calorie restriction does not elicit a robust extension of replicative lifespan inSaccharomyces cerevisiae", Proceedings of the National Academy of Sciences, vol. 111, pp. 11727-11731, 2014. http://dx.doi.org/10.1073/pnas.1410024111
- M.C. Jo, W. Liu, L. Gu, W. Dang, and L. Qin, "High-throughput analysis of yeast replicative aging using a microfluidic system", Proceedings of the National Academy of Sciences, vol. 112, pp. 9364-9369, 2015. http://dx.doi.org/10.1073/pnas.1510328112
- J.R. Delaney, A. Chou, B. Olsen, D. Carr, C. Murakami, U. Ahmed, S. Sim, E.H. An, A.S. Castanza, M. Fletcher, S. Higgins, M. Holmberg, J. Hui, M. Jelic, K. Jeong, J.R. Kim, S. Klum, E. Liao, M.S. Lin, W. Lo, H. Miller, R. Moller, Z.J. Peng, T. Pollard, P. Pradeep, D. Pruett, D. Rai, V. Ros, J. Schleit, A. Schuster, M. Singh, B.L. Spector, G.L. Sutphin, A.M. Wang, B.M. Wasko, H. Vander Wende, B.K. Kennedy, and M. Kaeberlein, "End-of-life cell cycle arrest contributes to stochasticity of yeast replicative aging", FEMS Yeast Research, vol. 13, pp. 267-276, 2013. http://dx.doi.org/10.1111/1567-1364.12030
- A. Dillin, D.E. Gottschling, and T. Nyström, "The good and the bad of being connected: the integrons of aging", Current Opinion in Cell Biology, vol. 26, pp. 107-112, 2014. http://dx.doi.org/10.1016/j.ceb.2013.12.003
- Y. Wei, H. Hu, G. Xie, N. Fu, Z. Ning, R. Zeng, and P. Khaitovich, "Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging", Genome Biology, vol. 16, 2015. http://dx.doi.org/10.1186/s13059-015-0608-2
- A. Ori, B. Toyama, M. Harris, T. Bock, M. Iskar, P. Bork, N. Ingolia, M. Hetzer, and M. Beck, "Integrated Transcriptome and Proteome Analyses Reveal Organ-Specific Proteome Deterioration in Old Rats", Cell Systems, vol. 1, pp. 224-237, 2015. http://dx.doi.org/10.1016/j.cels.2015.08.012
- K.A. Henderson, A.L. Hughes, and D.E. Gottschling, "Mother-daughter asymmetry of pH underlies aging and rejuvenation in yeast", eLife, vol. 3, 2014. http://dx.doi.org/10.7554/eLife.03504
COPYRIGHT
© 2016

Evidence for the hallmarks of human aging in replicatively aging yeast by Georges E. Janssens, Liesbeth M. Veenhoff is licensed under a Creative Commons Attribution 4.0 International License.








