
Reviews:
Microbial Cell, Vol. 4, No. 7, pp. 203 - 211; doi: 10.15698/mic2017.07.579
Impact of the host on Toxoplasma stage differentiation

1 Institute for Medical Microbiology, University Medical Center Goettingen, Goettingen, Germany.
Keywords: Toxoplasma gondii, stage conversion, bradyzoite, parasite host-interaction, host cell, metabolism, cell cycle, immune response.
Abbreviations:
HFF - human foreskin fibroblasts,
LDL - low-density lipoproteins,
LPS – lipopolysaccharide,
MyHC - myosin heavy chain,
NO – nitric oxide,
PV - parasitophorous vacuole,
SkMCs - skeletal muscle cells,
SNP - sodium nitroprusside.
Received originally: 20/04/2017 Received in revised form: 07/06/2017
Accepted: 07/06/2017
Published: 22/06/2017
Correspondence:
Carsten G.K. Lüder, Institute for Medical Microbiology, Kreuzbergring 57, 37075 Goettingen, Germany; Tel: +49 551 395869 clueder@gwdg.de
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Carsten G.K. Lüder and Taibur Rahman (2017). Impact of the host on Toxoplasma stage differentiation. Microbial Cell 4(7): 203-211. doi: 10.15698/mic2017.07.579
Abstract
The unicellular parasite Toxoplasma gondii infects warm-blooded animals and humans, and it is highly prevalent throughout the world. Infection of immunocompetent hosts is usually asymptomatic or benign but leads to long-term parasite persistence mainly within neural and muscular tissues. The transition from acute primary infection towards chronic toxoplasmosis is accompanied by a developmental switch from fast replicating and metabolically highly active tachyzoites to slow replicating and largely dormant bradyzoites within tissue cysts. Such developmental differentiation is critical for T. gondii in order to complete its life cycle and for pathogenesis. Herein, we summarize accumulating evidence indicating a major impact of the host cell physiology on stage conversion between the tachyzoite and the bradyzoite stage of the parasite. Withdrawal from cell cycle progression, proinflammatory responses, reduced availability of nutrients and extracellular adenosine can indeed induce tachyzoite-to-bradyzoite differentiation and tissue cyst formation. In contrast, high glycolytic activity as indicated by increased lactate secretion can inhibit bradyzoite formation. These examples argue for the intriguing possibility that after dissemination within its host, T. gondii can sense its cellular microenvironment to initiate the developmental program towards the bradyzoite stage in distinct cells. This may also explain the predominant localization of T. gondii in neural and muscular tissues during chronic toxoplasmosis.
INTRODUCTION
Developmental switching between life cycle stages is critical for the biology of a large number of unicellular microorganisms. It is also important for the course of infectious diseases caused by bacteria, fungi and protozoa. Examples include differentiation between planktonic and sessile bacteria during biofilm formation [1], between yeast and hyphae and vice versa during infection with distinct Candida spp. [2], or between Anopheles-transmitted sporozoites and liver stage merozoites of Plasmodium spp. during malaria [3]. Characterization of those factors that regulate these developmental processes is of fundamental interest to understand the biology of microbial cells and the evolution of multicellular organisms. It can also lead to the identification of novel targets for efficient intervening with onset or progression of infectious diseases.
–
Toxoplasma gondii is an obligate intracellular parasite of the phylum Apicomplexa which comprises a variety of pathogens of utmost importance for human and animal health. T. gondii is widespread throughout the world and threatens the health particularly of immunocompromised individuals or that of fetuses from recently infected pregnant women [4]. In otherwise healthy adults, primary infection with T. gondii is regularly controlled by the ensuing Th1-type cell-mediated immune response but nevertheless gives rise to chronic persistence of the parasite, possibly for the host’s life. The parasite’s ability to persist for extended periods of time is critical for transmission between hosts. Its appropriate regulation likely was and still is subject of selective pressure during evolution.
–
The life cycle of T. gondii is complex with members of the Felidae and warm-blooded vertebrates, i.e., mammals and birds, as final or intermediate hosts, respectively, and different phases of sexual and asexual reproduction. Not surprisingly, it also involves different parasite stages with sexual gametocytes, transmissible sporozoites, rapidly proliferating tachyzoites and dormant bradyzoites being the most prominent ones. Transition between the different stages requires extensive morphological remodeling, expression of distinct transcriptomes and proteomes, as well as several physiological adaptations [5][6]. The differentiation of tachyzoites to bradyzoites within intermediate hosts is experimentally amenable after in vitro infection of different host cells and after in vivo infection of laboratory or farm animals [5][7][8][9][10]. It is also of major biological and medical interest because the transition between tachyzoites and bradyzoites correlates with the acute or chronic phase of toxoplasmosis, respectively.
–
Tachyzoites of T. gondii develop after oral transmission of infective sporozoites or bradyzoites to intermediate hosts, presumably within the intestinal epithelium (Fig. 1). They rapidly divide within a membrane-bound intracellular compartment, i.e. the parasitophorous vacuole (PV) by a process called endodyogeny [11]. The doubling time of this stage depends on the parasite strain and can be as fast as ~5 hours in type I strains but takes 6 – 12 hours in type II and III strains [12]. Tachyzoites are metabolically highly active indicating a high demand for nutrients from the environment. After 5 to 6 cell divisions, tachyzoites egress from the host cell in a Ca2+-dependent manner and can then infect neighboring host cells [13][14]. T. gondii invades its host cell by a parasite-driven process that depends on the parasites’ actin-myosin machinery and the discharge of microneme and rhoptry proteins from the apical complex [15]. Importantly, the active host cell infection enables T. gondii to invade any nucleated cell of its host. Infection of monocytes and dendritic cells particularly contributes to the dissemination of the parasite throughout the hosts’ body [16][17]. A sustained parasite replication with tissue damage due to host cell lysis and/or the ensuing cell-mediated immune response correlates with the acute phase of infection and possibly overt disease under certain circumstances. In most cases, however, vigorous cell-mediated immunity is able to restrict tachyzoite replication and to even kill tachyzoites in a timely fashion thereby preventing overt symptoms.
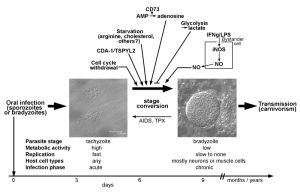 | FIGURE 1: T. gondii stage conversion and its regulation by the host (cell) microenvironment. Within one to two weeks of infection, highly active tachyzoites (two parasitophorous vacuoles within human foreskin fibroblasts are depicted; left micrograph) convert to relatively dormant bradyzoites (a tissue cyst from mouse brain containing hundreds of individual bradyzoites is shown; right micrograph). Formation of long-lived bradyzoite-containing tissue cysts is crucial for parasite transmission to new hosts. Reconversion of bradyzoites to tachyzoites can occur in immunocompromised individuals (e.g. those with AIDS or transplant recipients (TPX)) leading to life-threatening disease. Distinct characteristics of the infected host cell or of bystander cells can trigger or inhibit differentiation towards the bradyzoite stage as indicated in the upper part of the figure. Noteworthy, parasite-intrinsic triggers may also govern stage differentiation (not depicted). See main text for further details. |
–
Bradyzoites of T. gondii differentiate from tachyzoites within 6 – 9 days after oral infection of mice with oocysts or bradyzoites (Fig. 1) [11], but they can develop as early as 3 days after infection with tachyzoites [18]. Bradyzoites are also located inside a PV which, however, over time matures to a so-called intracellular tissue cyst. The most prominent feature of tissue cysts is the development of a thick cyst wall that is thought to confer structural rigidity [19]. Tissue cysts may contain several hundreds of bradyzoites which are generally slowly to non-replicating and metabolically rather inactive [5][11], but which can show episodic growth and replication [20]. Tissue cysts are long-lived although recent evidence suggests that they rupture at low but continuous frequency [21][22]. They are preferentially located in brain and muscular tissue, but they can be found in other organs as for instance liver and kidneys as well [11]. Tissue cysts persist for extended periods of time and are hence ideal for transmission of T. gondii to new hosts via carnivorism. Re-differentiation of bradyzoites to fast-replicating tachyzoites in immunocompromized patients, i.e. those with AIDS, leads to life-threatening reactivated toxoplasmosis, mostly presenting as Toxoplasma encephalitis [4].
–
Stage conversion between tachyzoites and bradyzoites is an area of intensive research. Both stages have been extensively characterized at the morphological and molecular level (reviewed in [6][11]). Additionally, important mechanisms of its developmental biology have been discovered within the last decade [23]. What remain less clear is how stage conversion is initiated and which role the host plays in this process. Answering this question may provide clues also for another intriguing issue; that is why tissue cysts are predominantly located in neuronal and muscular tissues. In this review we are summarizing recent evidences that the host cell physiology may determine whether T. gondii starts to differentiate from the tachyzoite to the bradyzoite stage or not.
HOST CELL FACTORS REGULATING STAGE CONVERSION
Cell type
Despite the ability of T. gondii to infect any nucleated cell of intermediate hosts, tissue cysts during chronic toxoplasmosis are not randomly distributed. The preferred localization appears to differ between host species [11]. They nonetheless show a predilection for neural and muscle tissues, including brain, eye, skeletal muscle and heart. It must be stressed, however, that T. gondii tissue cysts can develop in many different organs including liver and kidney. Within the brain, neurons are the main cell type that harbor tissue cysts during chronic toxoplasmosis [24][25][26]. The preferred cell type of tissue cyst development within other organs in vivo has not yet been determined.
–
Two main hypotheses have been proposed to explain the predominant localization in neural and muscular tissues; these are not mutually exclusive. According to the ‘immune privilege-based survival’ hypothesis bradyzoite-containing tissue cysts randomly form in (almost) all parasitized host cell types, but are subsequently eradicated in most tissues except immune-privileged organs including brain, eye and muscle tissues [27]. Whereas muscle tissue is not considered a classical immune-privileged organ, skeletal muscle cells (SkMCs) nonetheless display several characteristics of reduced immune functions under physiological conditions [28]. The ‘cell type-specific triggering’ hypothesis proposes that bradyzoite differentiation and tissue cyst formation are predominantly triggered in neural and muscular cells thus explaining the predilection for these tissues [29].
–
Neurons and mature skeletal muscle cells, i.e. myotubes, are post-mitotic, highly differentiated and long-living cells that cannot reenter productive cell cycle [30][31][32]. They therefore appear well suited to serve as host cells for a latent parasite stage that needs to survive for extended periods of time. Reasons for a predominant interaction of T. gondii with neurons in the brain came recently from elegant analyses of Cre reporter mice infected with Cre-secreting and fluorescently labelled parasites [26]. The results suggest that the higher frequency of neurons in the brain as compared to astrocytes and their larger cell surface favor infection of neurons rather than astrocytes. In addition, activation of astrocytes by IFN-γ further reduces the number of infected astrocytes either by killing of intracellular parasites or by preventing initial Toxoplasma-astrocyte interactions [26]. An impact of IFN-γ-dependent signaling in astrocytes on the distribution of tissue cysts was recently confirmed using mice with a specific depletion of STAT1 in astrocytes [33]. In contrast to astrocytes which are able to kill intracellular parasites in an IFN-γ-dependent fashion [34][35], neurons do not restrict T. gondii replication after activation with IFN-γ and/or TNF in vitro [36]. Thus, the predominant localization of T. gondii tissue cysts in neurons may result from the likelihood to infect this cell type and the absence of anti-parasitic effector mechanisms.
–
In addition, distinct cell types are more suitable to sustain tissue cyst formation after infection than others. Neurons and SkMCs readily sustain bradyzoite differentiation in vitro without the need of applying exogenous stress [29][37][38]. Intriguingly, not only the cell type but rather the state of the host cell appears to be critical. Using mouse SkMCs we recently showed that T. gondii readily differentiates to the bradyzoite stage in mature, myosin heavy chain (MyHC)-positive, syncytial myotubes, but not in proliferating, MyHC-negative precursors, i.e. in myoblasts or in fibroblasts (Fig. 2) [39]. This strongly suggests that the cellular microenvironment within differentiated myotubes may trigger a developmental pathway of T. gondii that leads to conversion to the persistent parasite stage. We thus propose that the ability of SkMCs to terminally differentiate rather than cell type characteristic(s) per se regulates the preferred localization of T. gondii tissue cysts in skeletal muscles. It is appealing to assume that terminal differentiation of neurons also triggers bradyzoite development, but this needs future experimental proof. Since myoblasts and myotubes differ in the expression of ~6.500 genes (Rahman & Lüder, unpublished), identification of host molecules/pathways triggering stage conversion in T. gondii is a challenge, but relevant factors are beginning to emerge (see below).
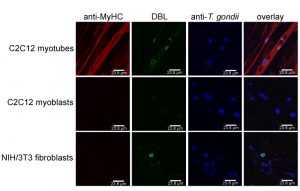 | FIGURE 2: T. gondii tissue cyst formation is supported by mature syncytial myotubes but not by proliferating myoblasts or fibroblasts. C2C12 murine skeletal muscle cells (SkMCs) were differentiated in vitro to myotubes. Myotubes, myoblasts and fibroblasts were infected with T. gondii for 72 hours. Efficient myotube formation was verified by staining with an antibody against myosin heavy chain (MyHC), a marker of differentiation to mature SkMCs (red fluorescence). Total parasites irrespective of the parasite stage were labelled with an antiserum (blue fluorescence) and T. gondii tissue cysts were labelled with Dilichos biflorus lectin (DBL) recognizing the tissue cyst wall (green fluorescence). Representative images were recorded by confocal laser scanning microscopy. |
–
Cell cycle
First evidence that the host cell cycle may regulate stage conversion in T. gondii came from observations that forced expression of the human cell division autoantigen (CDA)-1 promotes bradyzoite formation in type II and III strains (Fig. 1) [40]. CDA-1 (also known as testis-specific Y-encoded-like 2 (TSPYL2) or differentially expressed nucleolar transforming growth factor-beta 1 target (DENTT)) inhibits cell growth and proliferation by inducing the cell cycle inhibitor p21Waf1/Cip1 in a p53- and ERK1/2-dependent manner [41][42]. Pharmacological induction of CDA-1 in human-derived fibroblasts using a trisubstituted pyrrole (designated compound 1) or its ectopic expression in epithelial cells indeed inhibits parasite division and induces T. gondii bradyzoite antigen (BAG)-1 expression and formation of a cyst wall [40]. Consequently, knock-down of CDA-1 prevents the compound 1-induced reduction in parasite division and abolishes BAG-1 expression. Reduced parasite replication [43], expression of stage-specifically expressed BAG-1 (also designated BAG-5) [44][45] and formation of a cyst wall [11] are characteristics of the bradyzoite stage of T. gondii. Thus, the above findings indicate that host CDA-1 does suffice to trigger stage conversion in T. gondii. Importantly, CDA-1 has recently been proved to play a role in bradyzoite differentiation also in a more physiological environment of T. gondii stage conversion [39]. First, these authors provided direct evidence that the host cell cycle impacts T. gondii development. Thus, differentiation of the murine myoblast cell line C2C12 to polynucleated mature myotubes (see above) is accompanied by up-regulation of mRNA for CDA-1/TSPYL2, and by a halt in host cell cycle progression. As observed in mature SkMCs in vitro and in vivo, such differentiation and withdrawal from the host cell cycle induce T. gondii to convert from the tachyzoite to the bradyzoite stage (Fig. 1; see also above). Remarkably, shRNA knock-down of CDA-1/TSPYL2 in C2C12 SkMCs abolishes the increased bradyzoite formation as observed in mature myotubes and allows vigorous parasite replication [39]. It must be stressed that knock-down of CDA-1/TSPYL2 also prevents formation of myotubes suggesting multiple effects on the host cell including sustained cell cycle progression, inhibition of muscle cell differentiation and others. It nevertheless clearly links the withdrawal from the host cell cycle to stage differentiation in T. gondii. CDA-1/TSPYL2 expression and permanent arrest of mature SkMCs in a G0 phase of the cell cycle could thus provide an appropriate signal in muscle tissue in vivo to trigger T. gondii stage conversion. Intriguingly, in mice, CDA-1/TSPYL2 is highly expressed in different regions of the brain, including cerebral cortex and hippocampus and it is expressed to a lower extent also in the gonads [46][47]. It will thus be interesting to see whether CDA-1/TSPYL2 and cell cycle inhibition also triggers T. gondii stage conversion in the brain. The mechanism of how CDA-1 regulates bradyzoite differentiation is currently unknown.
–
Pro-inflammatory immune responses
Pro-inflammatory molecules were among the first host-derived signals for which pro-bradyzoite activities have been confirmed. Activation of murine macrophages with IFN-γ and/or LPS efficiently induces differentiation of T. gondii to the bradyzoite stage (Fig. 1) [48][49]. The level of macrophage activation is critical, since only intermediate levels of activation induce expression of bradyzoite-specific antigens [49]. This was explained by findings that only parasites that show limited replication but not a complete halt of replication are able to differentiate towards the bradyzoite stage [49]. Later on it was confirmed that transition through a G2-related cell cycle phase prior to mitosis and cytokinesis is required for T. gondii to enter the differentiation program to the bradyzoite stage [43]. Thus, non-activated host macrophages favor direct S phase-to-mitosis progression indicative for the tachyzoite cell cycle [12], while too strongly activated macrophages prevent T. gondii reaching a putative G2-associated checkpoint that may be required for initiating the developmental program towards bradyzoite formation. Bradyzoite formation in activated macrophages correlates with nitric oxide (NO) production by the inducible NO synthase (iNOS, also designated NOS2), and the NO donor sodium nitroprusside (SNP) partially mimics activation by IFN-γ/LPS [49]. The underlying mechanism of NO-mediated stage conversion in T. gondii seems to partially involve NO reacting with iron-sulfur centers of proteins of the host cells mitochondrial electron transport chain [49]. It is interesting to note that this links macrophage activation and its impact on bradyzoite formation in T. gondii with host cell metabolism (see also below). Since SNP even at high concentrations, however, only partially mimics the effect of macrophage activation, mechanisms others than the direct toxicity of NO appear to also impact bradyzoite formation. Induction of reactive oxygen species and starvation of T. gondii for tryptophan by activation of the oxidative burst [50] or the indoleamine 2,3-dioxygenase (IDO) [51][52], respectively, are possible candidates, but this needs to be validated. Furthermore, since iNOS catalyzes the production of NO by converting arginine to citrulline, and since T. gondii is auxotrophic for arginine [53][54], arginine limitation can also contribute to inducing T. gondii stage conversion in activated macrophages.
–
Other cell types including human foreskin fibroblasts (HFF), murine astrocytes or rat neurons, astrocytes and microglia do not increasingly support the differentiation from tachyzoites to bradyzoites after activation with IFN-γ [37][40][55][56][57]. This might be due to inappropriate activation of iNOS in these cells since SNP-derived exogenous NO does induce bradyzoite formation in human fibroblasts or rat brain cells to various extents [37][58][59]. In human fibroblasts, IL-6 also induces differentiation towards bradyzoites and formation of cyst-like structures [55]. Together, these data indicate that pro-inflammatory signals from the host cell can trigger stage conversion in T. gondii in a cell type- and context-dependent fashion. The significance of these findings for the course of infection in vivo is less clear. Detection of bradyzoites in vivo coincides with development of a vigorous cell-mediated immune response and this has supported the view that pro-inflammatory signals may trigger stage conversion in the infected host. Additionally, immunological competence is clearly required to prevent reactivation of latent tissue cysts during chronic toxoplasmosis in humans [4][60] and mice [61][62][63]. However, those cell types supporting bradyzoite formation after activation with pro-inflammatory cytokines are not preferred host cells for parasite persistence in vivo, and those which harbor latent tissue cysts during chronic infection do obviously not support bradyzoite formation in response to pro-inflammatory signals in vitro. Direct evidence for activation of host cells by pro-inflammatory molecules triggering stage conversion in vivo is thus lacking.
–
A possible scenario is, however, a bystander effect of activated inflammatory macrophages or other immune cells triggering stage conversion in adjacent Toxoplasma-infected non-hematopoietic cells including neurons and muscle cells via diffusion of NO (Fig. 1). Alternatively, activated macrophages could also trigger formation of bradyzoite-containing tissue cysts which can then infect neighboring cells via migration of free bradyzoites or even of tissue cysts [5]. Both scenarios await nevertheless future verification. It is nevertheless generally accepted that cell-mediated immunity is necessary to stabilize tissue cysts and to prevent reactivation [4][60][61]. This is supported by the finding that murine astrocytes support long-term cultivation of tissue cysts in vitro [57].
–
Host stress response
Applying extracellular stress including alkaline pH, heat shock or treatment of infected host cells with toxic substances as for instance sodium arsenite or mitochondrial inhibitors can induce bradyzoite differentiation and/or tissue cyst formation in vitro (reviewed in [29]). Whereas these stressors are artificial they have nonetheless helped to study T. gondii stage differentiation in vitro. In the context of this review their ability to induce stage differentiation raises the question whether they directly act on the parasite or indirectly via the host stress response or both. Exposure of extracellular parasites to alkaline pH or to SNP prior to infection of host cells in the absence of exogenous stress triggers bradyzoite development [58]. However, the extent to which bradyzoite formation is induced under these conditions is lower as compared to treatment of infected host cells with these stressors [58]. Whereas it is difficult to draw robust conclusions from these data due to different length of treatments, they nevertheless might suggest that stage conversion in T. gondii is both a direct parasite response to stress and an indirect effect mediated by the host cell. Whether or not a host stress response can trigger stage differentiation in vivo awaits clarification.
–
Metabolic features
The fast replicating tachyzoites rely on higher nutrient uptake from the host cell than the more quiescent bradyzoites in order to satisfy their demands for energy and metabolic building blocks. T. gondii is auxotrophic for several metabolites including purines, arginine, cysteine [64], those amino acids that are also essential for humans except lysine [64], polyamines, cholesterol, choline and vitamins [53][54]. Limited supply of essential molecules and basic nutrients from the host cell may thus restrict tachyzoite replication and thereby favor developmental switching towards the bradyzoite stage (Fig. 1) [43][49]. This view has been convincingly confirmed when intracellular parasites were grown in culture medium containing not more than 5-10 µM of arginine [65]. It is remarkable that arginine starvation triggers stage conversion to high extent even in a parasite strain that is generally rather refractory to tissue cyst formation (RH strain). However, arginine plasma concentrations in mammals including humans vary between 100 and 250 µM [66] and thus exceed by far those 5-10 µM required to trigger stage conversion in T. gondii. In addition, the reported intracellular arginine concentrations of 100 to 1000 µM in human cells [66] are also comparatively high. The intracellular threshold necessary to trigger stage differentiation in T. gondii is unknown, but these figures make it rather unlikely that arginine can drop in vivo to levels that suffice to trigger bradyzoite formation. Starvation of T. gondii for multiple nutrients might however suffice to favor stage differentiation. For instance, depletion of lipoproteins from culture medium also induces stage conversion which is abolished by addition of low-density lipoproteins (LDL) [67]. Since LDL particles are major cholesterol carriers in the blood, and since T. gondii is auxotrophic for LDL-derived cholesterol [68], the authors concluded that starvation of LDL-derived cholesterol can indeed trigger bradyzoite formation [67]. Whether limited availability of LDL-cholesterol regulates stage conversion in vivo needs experimental validation. It is noteworthy that infection of Chinese hamster ovary cells with T. gondii tachyzoites increases LDL uptake by threefold [68] suggesting a particularly high demand for cholesterol of this parasite stage that cannot be satisfied by the regular cholesterol uptake of these cells.
–
Ambient CO2 concentration (0.03%) in the environment triggers bradyzoite formation and tissue cyst formation in a parasite strain that is prone to stage conversion in vitro and in vivo (Prugniaud strain) [5]. T. gondii predominantly synthesizes pyrimidines de novo from amino acids, bicarbonate and 5-phosphoribosyl-1-pyrophosphate [53]. Low CO2/bicarbonate may therefore limit the availability of pyrimidines and hence inhibit DNA and RNA synthesis by T. gondii. Due to CO2 concentrations in human blood between 4.5 to 5.8% at standard atmospheric pressure it is however unlikely that CO2/bicarbonate availability is a trigger of stage conversion under physiological conditions.
–
Contrary to the examples above where starvation of T. gondii triggers bradyzoite formation, distinct metabolites the parasite needs for optimal growth can also trigger bradyzoite formation. The 5’-ecto-nucleosidase (CD73) is expressed on the surface of various mammalian cells including CNS cells and dephosphorylates adenosine monophosphate (AMP) to adenosine. This in turn dampens excessive inflammation via adenosine receptor signaling or can be used for purine salvage. T. gondii is unable to synthesize purines de novo (see above) [53][54] and adenosine is its major source for purines [69]. Intriguingly, CD73-deficient mice show reduced bradyzoite differentiation and reduced tissue cyst burden in their brains (Fig. 1) and consequently a decreased mortality as compared to wild type mice [70]. Reduced bradyzoite and tissue cyst formation in stressed CD73-/- astrocytes can be rescued with exogenous adenosine. Furthermore, adenosine receptor signaling is dispensable for triggering stage differentiation by adenosine. Thus, external adenosine can trigger stage differentiation in T. gondii probably after its uptake from the environment. Whether bradyzoites have an unexpectedly high demand for adenosine or whether it functions as a signaling molecule to trigger stage conversion awaits future clarification.
–
A link between lactate production by different host cells and differentiation of T. gondii has been established [71]. Indeed, increased glycolytic activity as evidenced by secretion of lactate correlates with the inability of NIH/3T3 fibroblasts and 293T embryonic kidney cells to sustain bradyzoite formation under stress conditions. Supernatants from these cells inhibit bradyzoite formation also in HFF fibroblasts and Vero kidney cells which are normally permissive for bradyzoite formation. Furthermore, forced activation of glycolysis in HFF and Vero cells by expression of a myristoylated, i.e. an active version of the Akt kinase and/or by increasing glucose levels also inhibit stage conversion [71]. The data thus suggest that high glycolytic activity of the host cell provides nutrients to the parasites that suffice to sustain the high metabolic demands of tachyzoites thereby inhibiting bradyzoite formation (Fig. 1). They also provide compelling evidence that metabolic features of the host cell can indeed impact stage conversion in T. gondii. It must be stressed that lactate itself accounts for only 30% of the inhibitory activity of 293T supernatant and that it only inhibits bradyzoite formation in Vero cells but not in HFF cells [71]. Thus, HFF and Vero cells obviously differ in their regulation of glycolytic activity by external lactate. It is also important to note that increased glycolysis only prevents stress-induced stage conversion. Whether it can also inhibit spontaneous stage conversion as observed in distinct cell types (see above), or whether low glycolytic activity of the host cell does suffice to facilitate stage conversion in distinct cells needs to be clarified.
–
Collectively, these data provide clear evidence that distinct metabolic characteristics of the host cell can regulate stage conversion in T. gondii.
CONCLUSIONS
The data discussed above provide strong evidence that the host cell microenvironment regulates stage differentiation in T. gondii. Host cell cycle, host cell metabolism and inflammatory responses, the latter possibly by acting on infected non-immune bystander cells, clearly affect bradyzoite formation in vitro. Distinct features as for instance the CD73-mediated adenosine formation have been proved to regulate bradyzoite and tissue cyst formation also in vivo (see above). We thus propose that differentiation towards the bradyzoite stage can occur spontaneously when the parasite encounters an appropriate host cell environment. Terminally differentiated SkMCs and neurons may represent such a suitable cellular niche and this may explain the predominant localization of tissue cysts in muscular and neural tissues during chronic toxoplasmosis. Inflammatory responses including NO production may additionally limit parasite replication and hence promote stage differentiation. Following infection with sporozoites or bradyzoites, T. gondii can also spontaneously slow down its cell cycle and differentiate to the bradyzoite stage after ~20 divisions of rapid tachyzoite growth, i.e. via a programmed developmental pathway [43][72]. The factors regulating tachyzoite-to-bradyzoite differentiation are thus likely diverse and complex.
–
From the findings discussed in this review several important questions emerge. Firstly, do host cell cycle, host cell metabolism and inflammatory responses act independently on T. gondii to initiate bradyzoite differentiation? Secondly, what are the host cell signals that are perceived by the parasite and how are these transduced into the program that ultimately leads to stage conversion? And finally, do the same signals trigger stage conversion within different host cell types (and/or different host species) and does this indeed explain the predilection of T. gondii to persist in certain tissues, i.e. brain and muscle? Elucidating these issues is critical to better understand the developmental processes in T. gondii and possibly also in other Apicomplexa.
References
- D. Claessen, D.E. Rozen, O.P. Kuipers, L. Søgaard-Andersen, and G.P. van Wezel, "Bacterial solutions to multicellularity: a tale of biofilms, filaments and fruiting bodies", Nature Reviews Microbiology, vol. 12, pp. 115-124, 2014. http://dx.doi.org/10.1038/nrmicro3178
- D. Hnisz, A.F. Bardet, C.J. Nobile, A. Petryshyn, W. Glaser, U. Schöck, A. Stark, and K. Kuchler, "A Histone Deacetylase Adjusts Transcription Kinetics at Coding Sequences during Candida albicans Morphogenesis", PLoS Genetics, vol. 8, pp. e1003118, 2012. http://dx.doi.org/10.1371/journal.pgen.1003118
- I. Coppens, "Metamorphoses of malaria: the role of autophagy in parasite differentiation", Essays in Biochemistry, vol. 51, pp. 127-136, 2011. http://dx.doi.org/10.1042/bse0510127
- J. Montoya, and O. Liesenfeld, "Toxoplasmosis", The Lancet, vol. 363, pp. 1965-1976, 2004. http://dx.doi.org/10.1016/S0140-6736(04)16412-X
- F. Dzierszinski, M. Nishi, L. Ouko, and D.S. Roos, "Dynamics of Toxoplasma gondii Differentiation", Eukaryotic Cell, vol. 3, pp. 992-1003, 2004. http://dx.doi.org/10.1128/EC.3.4.992-1003.2004
- L.J. Knoll, T. Tomita, and L.M. Weiss, "Bradyzoite Development", Toxoplasma Gondii, pp. 521-549, 2014. http://dx.doi.org/10.1016/B978-0-12-396481-6.00015-5
- D.S. Lindsay, J.P. Dubey, B.L. Blagburn, and M. Toivio-Kinnucan, "Examination of Tissue Cyst Formation by Toxoplasma gondii in Cell Cultures Using Bradyzoites, Tachyzoites, and Sporozoites", The Journal of Parasitology, vol. 77, pp. 126, 1991. http://dx.doi.org/10.2307/3282569
- T. McHugh, R. Holliman, and P. Butcher, "The in vitro model of tissue cyst formation in Toxoplasma gondii", Parasitology Today, vol. 10, pp. 281-285, 1994. http://dx.doi.org/10.1016/0169-4758(94)90148-1
- D. Ferguson, "Use of molecular and ultrastructural markers to evaluate stage conversion of Toxoplasma gondii in both the intermediate and definitive host", International Journal for Parasitology, vol. 34, pp. 347-360, 2004. http://dx.doi.org/10.1016/j.ijpara.2003.11.024
- K.J. Pittman, M.T. Aliota, and L.J. Knoll, "Dual transcriptional profiling of mice and Toxoplasma gondii during acute and chronic infection", BMC Genomics, vol. 15, 2014. http://dx.doi.org/10.1186/1471-2164-15-806
- J.P. Dubey, D.S. Lindsay, and C.A. Speer, "Structures of Toxoplasma gondii tachyzoites, bradyzoites, and sporozoites and biology and development of tissue cysts.", Clinical microbiology reviews, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9564564
- J. Radke, "Defining the cell cycle for the tachyzoite stage of Toxoplasma gondii", Molecular and Biochemical Parasitology, vol. 115, pp. 165-175, 2001. http://dx.doi.org/10.1016/S0166-6851(01)00284-5
- I.J. Blader, B.I. Coleman, C. Chen, and M. Gubbels, "Lytic Cycle of Toxoplasma gondii: 15 Years Later", Annual Review of Microbiology, vol. 69, pp. 463-485, 2015. http://dx.doi.org/10.1146/annurev-micro-091014-104100
- K. Nagamune, L.M. Hicks, B. Fux, F. Brossier, E.N. Chini, and L.D. Sibley, "Abscisic acid controls calcium-dependent egress and development in Toxoplasma gondii", Nature, vol. 451, pp. 207-210, 2008. http://dx.doi.org/10.1038/nature06478
- K. Frénal, V. Polonais, J. Marq, R. Stratmann, J. Limenitakis, and D. Soldati-Favre, "Functional Dissection of the Apicomplexan Glideosome Molecular Architecture", Cell Host & Microbe, vol. 8, pp. 343-357, 2010. http://dx.doi.org/10.1016/j.chom.2010.09.002
- H. Lambert, N. Hitziger, I. Dellacasa, M. Svensson, and A. Barragan, "Induction of dendritic cell migration upon Toxoplasma gondii infection potentiates parasite dissemination", Cellular Microbiology, vol. 8, pp. 1611-1623, 2006. http://dx.doi.org/10.1111/j.1462-5822.2006.00735.x
- N. Courret, S. Darche, P. Sonigo, G. Milon, D. Buzoni-Gâtel, and I. Tardieux, "CD11c- and CD11b-expressing mouse leukocytes transport single Toxoplasma gondii tachyzoites to the brain", Blood, vol. 107, pp. 309-316, 2006. http://dx.doi.org/10.1182/blood-2005-02-0666
- J.P. DUBEY, and J.K. FRENKEL, "Feline Toxoplasmosis from Acutely Infected Mice and the Development of Toxoplasma Cysts*", The Journal of Protozoology, vol. 23, pp. 537-546, 1976. http://dx.doi.org/10.1111/j.1550-7408.1976.tb03836.x
- T. Tomita, D.J. Bzik, Y.F. Ma, B.A. Fox, L.M. Markillie, R.C. Taylor, K. Kim, and L.M. Weiss, "The Toxoplasma gondii Cyst Wall Protein CST1 Is Critical for Cyst Wall Integrity and Promotes Bradyzoite Persistence", PLoS Pathogens, vol. 9, pp. e1003823, 2013. http://dx.doi.org/10.1371/journal.ppat.1003823
- E. Watts, Y. Zhao, A. Dhara, B. Eller, A. Patwardhan, and A.P. Sinai, "Novel Approaches Reveal that Toxoplasma gondii Bradyzoites within Tissue Cysts Are Dynamic and Replicating EntitiesIn Vivo", mBio, vol. 6, 2015. http://dx.doi.org/10.1128/mBio.01155-15
- D.J.P. Ferguson, W.M. Hutchison, and E. Pettersen, "Tissue cyst rupture in mice chronically infected withToxoplasma gondii", Parasitology Research, vol. 75, pp. 599-603, 1989. http://dx.doi.org/10.1007/bf00930955
- J.P. Dubey, L.R. Ferreira, M. Alsaad, S.K. Verma, D.A. Alves, G.N. Holland, and G.A. McConkey, "Experimental Toxoplasmosis in Rats Induced Orally with Eleven Strains of Toxoplasma gondii of Seven Genotypes: Tissue Tropism, Tissue Cyst Size, Neural Lesions, Tissue Cyst Rupture without Reactivation, and Ocular Lesions", PLOS ONE, vol. 11, pp. e0156255, 2016. http://dx.doi.org/10.1371/journal.pone.0156255
- M.W. White, J.R. Radke, and J.B. Radke, "Toxoplasmadevelopment - turn the switch on or off?", Cellular Microbiology, vol. 16, pp. 466-472, 2014. http://dx.doi.org/10.1111/cmi.12267
- D.J.P. Ferguson, and W.M. Hutchison, "An ultrastructural study of the early development and tissue cyst formation of Toxoplasma gondii in the brains of mice", Parasitology Research, vol. 73, pp. 483-491, 1987. http://dx.doi.org/10.1007/bf00535321
- D.J.P. Ferguson, and W.M. Hutchison, "The host-parasite relationship ofToxoplasma gondii in the brains of chronically infected mice", Virchows Archiv A Pathological Anatomy and Histopathology, vol. 411, pp. 39-43, 1987. http://dx.doi.org/10.1007/bf00734512
- C.M. Cabral, S. Tuladhar, H.K. Dietrich, E. Nguyen, W.R. MacDonald, T. Trivedi, A. Devineni, and A.A. Koshy, "Neurons are the Primary Target Cell for the Brain-Tropic Intracellular Parasite Toxoplasma gondii", PLOS Pathogens, vol. 12, pp. e1005447, 2016. http://dx.doi.org/10.1371/journal.ppat.1005447
- K. Kristensson, J.D.M. Mhlanga, and M. Bentivoglio, "Parasites and the Brain: Neuroinvasion, Immunopathogenesis and Neuronal Dysfunctions", Current Topics in Microbiology and Immunology, pp. 227-257, 2002. http://dx.doi.org/10.1007/978-3-662-09525-6_12
- H. Wiendl, R. Hohlfeld, and B.C. Kieseier, "Immunobiology of muscle: advances in understanding an immunological microenvironment", Trends in Immunology, vol. 26, pp. 373-380, 2005. http://dx.doi.org/10.1016/j.it.2005.05.003
- M.D.F. Ferreira da Silva, H.S. Barbosa, U. Groß, and C.G.K. Lüder, "Stress-related and spontaneous stage differentiation of Toxoplasma gondii", Molecular BioSystems, vol. 4, pp. 824, 2008. http://dx.doi.org/10.1039/b800520f
- K. Walsh, and H. Perlman, "Cell cycle exit upon myogenic differentiation", Current Opinion in Genetics & Development, vol. 7, pp. 597-602, 1997. http://dx.doi.org/10.1016/S0959-437X(97)80005-6
- E.S. Deneris, and O. Hobert, "Maintenance of postmitotic neuronal cell identity", Nature Neuroscience, vol. 17, pp. 899-907, 2014. http://dx.doi.org/10.1038/nn.3731
- J.M. Frade, and M.C. Ovejero-Benito, "Neuronal cell cycle: the neuron itself and its circumstances", Cell Cycle, vol. 14, pp. 712-720, 2015. http://dx.doi.org/10.1080/15384101.2015.1004937
- S. Hidano, L.M. Randall, L. Dawson, H.K. Dietrich, C. Konradt, P.J. Klover, B. John, T.H. Harris, Q. Fang, B. Turek, T. Kobayashi, L. Hennighausen, D.P. Beiting, A.A. Koshy, and C.A. Hunter, "STAT1 Signaling in Astrocytes Is Essential for Control of Infection in the Central Nervous System", mBio, vol. 7, 2016. http://dx.doi.org/10.1128/mBio.01881-16
- P.K. Peterson, G. Gekker, S. Hu, and C.C. Chao, "Human Astrocytes Inhibit Intracellular Multiplication of Toxoplasma gondii by a Nitric Oxide-Mediated Mechanism", Journal of Infectious Diseases, vol. 171, pp. 516-518, 1995. http://dx.doi.org/10.1093/infdis/171.2.516
- S.K. Halonen, G.A. Taylor, and L.M. Weiss, "Gamma Interferon-Induced Inhibition ofToxoplasma gondiiin Astrocytes Is Mediated by IGTP", Infection and Immunity, vol. 69, pp. 5573-5576, 2001. http://dx.doi.org/10.1128/iai.69.9.5573-5576.2001
- D. Schlüter, M. Deckert, H. Hof, and K. Frei, "Toxoplasma gondiiInfection of Neurons Induces Neuronal Cytokine and Chemokine Production, but Gamma Interferon- and Tumor Necrosis Factor-Stimulated Neurons Fail To Inhibit the Invasion and Growth ofT. gondii", Infection and Immunity, vol. 69, pp. 7889-7893, 2001. http://dx.doi.org/10.1128/IAI.69.12.7889-7893.2001
- C.G. Lüder, M. Giraldo-Velásquez, M. Sendtner, and U. Gross, "Toxoplasma gondii in Primary Rat CNS Cells: Differential Contribution of Neurons, Astrocytes, and Microglial Cells for the Intracerebral Development and Stage Differentiation", Experimental Parasitology, vol. 93, pp. 23-32, 1999. http://dx.doi.org/10.1006/expr.1999.4421
- M.D.F. Ferreira-da-Silva, A.C. Takács, H.S. Barbosa, U. Gross, and C.G. Lüder, "Primary skeletal muscle cells trigger spontaneous Toxoplasma gondii tachyzoite-to-bradyzoite conversion at higher rates than fibroblasts", International Journal of Medical Microbiology, vol. 299, pp. 381-388, 2009. http://dx.doi.org/10.1016/j.ijmm.2008.10.002
- I.J. Swierzy, and C.G.K. Lüder, "Withdrawal of skeletal muscle cells from cell cycle progression triggers differentiation ofToxoplasma gondiitowards the bradyzoite stage", Cellular Microbiology, vol. 17, pp. 2-17, 2014. http://dx.doi.org/10.1111/cmi.12342
- J.R. Radke, R.G. Donald, A. Eibs, M.E. Jerome, M.S. Behnke, P. Liberator, and M.W. White, "Changes in the Expression of Human Cell Division Autoantigen-1 Influence Toxoplasma gondii Growth and Development", PLoS Pathogens, vol. 2, pp. e105, 2006. http://dx.doi.org/10.1371/journal.ppat.0020105
- Z. Chai, B. Sarcevic, A. Mawson, and B. Toh, "SET-related Cell Division Autoantigen-1 (CDA1) Arrests Cell Growth", Journal of Biological Chemistry, vol. 276, pp. 33665-33674, 2001. http://dx.doi.org/10.1074/jbc.M007681200
- Y. Tu, W. Wu, T. Wu, Z. Cao, R. Wilkins, B. Toh, M.E. Cooper, and Z. Chai, "Antiproliferative Autoantigen CDA1 Transcriptionally Up-regulates p21Waf1/Cip1 by Activating p53 and MEK/ERK1/2 MAPK Pathways", Journal of Biological Chemistry, vol. 282, pp. 11722-11731, 2007. http://dx.doi.org/10.1074/jbc.M609623200
- J.R. Radke, M.N. Guerini, M. Jerome, and M.W. White, "A change in the premitotic period of the cell cycle is associated with bradyzoite differentiation in Toxoplasma gondii", Molecular and Biochemical Parasitology, vol. 131, pp. 119-127, 2003. http://dx.doi.org/10.1016/S0166-6851(03)00198-1
- W. Bohne, U. Gross, D.J.P. Ferguson, and J. Heesemann, "Cloning and characterization of a bradyzoite‐specifically expressed gene (hsp30/bag1) of Toxoplasma gondii, related to genes encoding small heat‐shock proteins of plants", Molecular Microbiology, vol. 16, pp. 1221-1230, 1995. http://dx.doi.org/10.1111/j.1365-2958.1995.tb02344.x
- S.F. Parmley, L.M. Weiss, and S. Yang, "Cloning of a bradyzoite-specific gene of Toxoplasma gondii encoding a cytoplasmic antigen", Molecular and Biochemical Parasitology, vol. 73, pp. 253-257, 1995. http://dx.doi.org/10.1016/0166-6851(95)00100-f
- C. Lin, T. Huang, G. Wang, T. Kuo, T. Yen, and Y. Hsueh, "Neural activity‐ and development‐dependent expression and distribution of CASK interacting nucleosome assembly protein in mouse brain", Journal of Comparative Neurology, vol. 494, pp. 606-619, 2005. http://dx.doi.org/10.1002/cne.20825
- K.P. Tao, S.W. Fong, Z. Lu, Y.P. Ching, K.W. Chan, and S.Y. Chan, "TSPYL2 Is Important for G1 Checkpoint Maintenance upon DNA Damage", PLoS ONE, vol. 6, pp. e21602, 2011. http://dx.doi.org/10.1371/journal.pone.0021602
- W. Bohne, J. Heesemann, and U. Gross, "Induction of bradyzoite-specific Toxoplasma gondii antigens in gamma interferon-treated mouse macrophages.", Infection and immunity, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8432596
- W. Bohne, J. Heesemann, and U. Gross, "Reduced replication of Toxoplasma gondii is necessary for induction of bradyzoite-specific antigens: a possible role for nitric oxide in triggering stage conversion.", Infection and immunity, 1994. http://www.ncbi.nlm.nih.gov/pubmed/8168938
- H.W. Murray, B.Y. Rubin, S.M. Carriero, A.M. Harris, and E.A. Jaffee, "Human mononuclear phagocyte antiprotozoal mechanisms: oxygen-dependent vs oxygen-independent activity against intracellular Toxoplasma gondii.", Journal of immunology (Baltimore, Md. : 1950), 1985. http://www.ncbi.nlm.nih.gov/pubmed/2981929
- A. Habara-Ohkubo, T. Shirahata, O. Takikawa, and R. Yoshida, "Establishment of an antitoxoplasma state by stable expression of mouse indoleamine 2,3-dioxygenase.", Infection and immunity, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8478070
- W. Däubener, and C.R. MacKenzie, "Ifn-γ Activated Indoleamine 2,3-Dioxygenase Activity in Human Cells is an Antiparasitic and an Antibacterial Effector Mechanism", Advances in Experimental Medicine and Biology, pp. 517-524, 1999. http://dx.doi.org/10.1007/978-1-4615-4709-9_64
- E.A. Innes, "", The Veterinary Journal, vol. 177, pp. 149-150, 2008. http://dx.doi.org/10.1016/j.tvjl.2008.02.017
- C.G.K. Lüder, and F. Seeber, "Toxoplasma", Molecular Parasitology, pp. 217-239, 2016. http://dx.doi.org/10.1007/978-3-7091-1416-2_8
- L.M. WEISS, D. LAPLACE, P.M. TAKVORIAN, H.B. TANOWITZ, A. CALI, and M. WITTNER, "A Cell Culture System for Study of the Development of Toxoplasma gondii Bradyzoites", Journal of Eukaryotic Microbiology, vol. 42, pp. 150-157, 1995. http://dx.doi.org/10.1111/j.1550-7408.1995.tb01556.x
- M. Soete, D. Camus, and J. Dubrametz, "Experimental Induction of Bradyzoite-Specific Antigen Expression and Cyst Formation by the RH Strain of Toxoplasma gondii in Vitro", Experimental Parasitology, vol. 78, pp. 361-370, 1994. http://dx.doi.org/10.1006/expr.1994.1039
- T.C. Jones, K.A. Bienz, and P. Erb, "In vitro cultivation of Toxoplasma gondii cysts in astrocytes in the presence of gamma interferon.", Infection and immunity, 1986. http://www.ncbi.nlm.nih.gov/pubmed/3079728
- L.M. Weiss, Y.F. Ma, P.M. Takvorian, H.B. Tanowitz, and M. Wittner, "Bradyzoite development in Toxoplasma gondii and the hsp70 stress response.", Infection and immunity, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9632598
- L.A. Kirkman, L.M. Weiss, and K. Kim, "Cyclic Nucleotide Signaling in Toxoplasma gondii Bradyzoite Differentiation", Infection and Immunity, vol. 69, pp. 148-153, 2001. http://dx.doi.org/10.1128/IAI.69.1.148-153.2001
- A. Mamidi, J.A. DeSimone, and R.J. Pomerantz, "Central nervous system infections in individuals with HIV-1 infection", Journal of Neurovirology, vol. 8, pp. 158-167, 2002. http://dx.doi.org/10.1080/13550280290049723
- R. Gazzinelli, Y. Xu, S. Hieny, A. Cheever, and A. Sher, "Simultaneous depletion of CD4+ and CD8+ T lymphocytes is required to reactivate chronic infection with Toxoplasma gondii.", Journal of immunology (Baltimore, Md. : 1950), 1992. http://www.ncbi.nlm.nih.gov/pubmed/1351500
- M.H. Beaman, F.G. Araujo, and J.S. Remington, "Protective Reconstitution of the SCID Mouse against Reactivation of Toxoplasmic Encephalitis", Journal of Infectious Diseases, vol. 169, pp. 375-383, 1994. http://dx.doi.org/10.1093/infdis/169.2.375
- T.M. Scharton-Kersten, G. Yap, J. Magram, and A. Sher, "Inducible Nitric Oxide Is Essential for Host Control of Persistent but Not Acute Infection with the Intracellular Pathogen Toxoplasma gondii", The Journal of Experimental Medicine, vol. 185, pp. 1261-1274, 1997. http://dx.doi.org/10.1084/jem.185.7.1261
- K. Chaudhary, and D.S. Roos, "Protozoan genomics for drug discovery", Nature Biotechnology, vol. 23, pp. 1089-1091, 2005. http://dx.doi.org/10.1038/nbt0905-1089
- B.A. Fox, J.P. Gigley, and D.J. Bzik, "Toxoplasma gondii lacks the enzymes required for de novo arginine biosynthesis and arginine starvation triggers cyst formation", International Journal for Parasitology, vol. 34, pp. 323-331, 2004. http://dx.doi.org/10.1016/j.ijpara.2003.12.001
- G. WU, and S.M. MORRIS, "Arginine metabolism: nitric oxide and beyond", Biochemical Journal, vol. 336, pp. 1-17, 1998. http://dx.doi.org/10.1042/bj3360001
- F. Ihara, and Y. Nishikawa, "Starvation of low-density lipoprotein-derived cholesterol induces bradyzoite conversion in Toxoplasma gondii", Parasites & Vectors, vol. 7, pp. 248, 2014. http://dx.doi.org/10.1186/1756-3305-7-248
- I. Coppens, A.P. Sinai, and K.A. Joiner, "Toxoplasma gondii Exploits Host Low-Density Lipoprotein Receptor-Mediated Endocytosis for Cholesterol Acquisition", The Journal of Cell Biology, vol. 149, pp. 167-180, 2000. http://dx.doi.org/10.1083/jcb.149.1.167
- E.C. Krug, J.J. Marr, and R.L. Berens, "Purine metabolism in Toxoplasma gondii.", The Journal of biological chemistry, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2732241
- D.A. Mahamed, J.H. Mills, C.E. Egan, E.Y. Denkers, and M.S. Bynoe, "CD73-generated adenosine facilitatesToxoplasma gondiidifferentiation to long-lived tissue cysts in the central nervous system", Proceedings of the National Academy of Sciences, vol. 109, pp. 16312-16317, 2012. http://dx.doi.org/10.1073/pnas.1205589109
- D.R. Weilhammer, A.T. Iavarone, E.N. Villegas, G.A. Brooks, A.P. Sinai, and W.C. Sha, "Host metabolism regulates growth and differentiation of Toxoplasma gondii", International Journal for Parasitology, vol. 42, pp. 947-959, 2012. http://dx.doi.org/10.1016/j.ijpara.2012.07.011
- M.E. Jerome, J.R. Radke, W. Bohne, D.S. Roos, and M.W. White, "Toxoplasma gondii bradyzoites form spontaneously during sporozoite-initiated development.", Infection and immunity, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9746587
ACKNOWLEDGMENTS
We are grateful to Izabela Swierzy for providing micrograph images presented in Figure 2. Taibur Rahman is supported by a PhD scholarship from Interweave Erasmus Mundus Action 2 Partnership. We also acknowledge support by the German Research Foundation (DFG) and the Open Access Publication Funds of the University of Goettingen.
COPYRIGHT
© 2017

Impact of the host on Toxoplasma stage differentiation by Lüder and Rahman is licensed under a Creative Commons Attribution 4.0 International License.








