
Research Articles:
Microbial Cell, Vol. 4, No. 6, pp. 182 - 190; doi: 10.15698/mic2017.06.576
Integrative modules for efficient genome engineering in yeast
1 Department of Cell and Developmental Biology, Hebrew University of Jerusalem, Givat Ram, Jerusalem 91904, Israel.
Keywords: vector, bidirectional promoter, integrative plasmid, genetic integration, yeast, Saccharomyces cerevisiae.
Received originally: 05/01/2017 Received in revised form: 23/05/2017
Accepted: 24/05/2017
Published: 05/06/2017
Correspondence:
Daniel Kaganovich, dan@cc.huji.ac.il
Triana Amen, amentri@gmail.com
Conflict of interest statement: The authors declare no conflicts of interest.
Please cite this article as: Triana Amen and Daniel Kaganovich (2017). Integrative modules for efficient genome engineering in yeast. Microbial Cell 4(6):182-190. doi: 10.15698/mic2017.06.576
Abstract
We present a set of vectors containing integrative modules for efficient genome integration into the commonly used selection marker loci of the yeast Saccharomyces cerevisiae. A fragment for genome integration is generated via PCR with a unique set of short primers and integrated into HIS3, URA3, ADE2, and TRP1 loci. The desired level of expression can be achieved by using constitutive (TEF1p, GPD1p), inducible (CUP1p, GAL1/10p), and daughter-specific (DSE4p) promoters available in the modules. The reduced size of the integrative module compared to conventional integrative plasmids allows efficient integration of multiple fragments. We demonstrate the efficiency of this tool by simultaneously tagging markers of the nucleus, vacuole, actin, and peroxisomes with genomically integrated fluorophores. Improved integration of our new pDK plasmid series allows stable introduction of several genes and can be used for multi-color imaging. New bidirectional promoters (TEF1p-GPD1p, TEF1p-CUP1p, and TEF1p-DSE4p) allow tractable metabolic engineering.
INTRODUCTION
Saccharomyces cerevisiae is an indispensable tool for high throughput studies of biological processes. The ever-increasing diversity of tools for yeast genome manipulation allows systematic examination of biological pathways in a highly physiological, cost-effective, and tractable manner [1][2][3][4][5][6][7]. However, attempts to introduce multiple genes into chromosomal loci often encounter complications due to decreased efficiency and cloning limitations, such as overlaps in restriction site usage amongst multiple inserts [7][8]. In order to overcome the difficulties of multiple gene integrations we sought to expand the variety of yeast integration cassettes with a specially designed pDK vector set.
–
Several methods enable rapid gene introduction. The most common ones are ectopic plasmid expression and chromosomal integration. Ectopic expression via multicopy or centromeric plasmids is often faster and easier than integration, but poses problems due to highly inhomogeneous expression [9][10]. Hence, multi-color imaging and metabolic engineering in yeast often require stable integration of genes. Genome integration via homologous recombination has advantages over ectopic expression: e. g. stable strains, controlled copy number, and uniform expression [9].
–
Several integration strategies are commonly employed. (1) Yeast integrative plasmids are introduced into the genome in linearized form [1]. The linearized fragment contains the desired insert and selection marker, but also substantial superfluous genetic material including the bacterial selection marker and replication origins [1]. These extraneous sequences may reduce integration efficiency [8]. Additionally, the customized insert must not contain the restriction site used for plasmid linearization, adding further limitations. (2) Another frequently used strategy is PCR amplification using extended primers with homology regions allowing integration into any locus. Since the primers provide relatively short homology regions, the integration efficiency is comparatively low with this approach as well [8]. Insertion into loci rather than selective markers does not pose a particular advantage since the selective marker locus still has to be present in the integrated fragment. (3) Recent advances in CRISPR/Cas9 genome engineering allow highly efficient introduction of multiple fragments with short homology regions and often without the need for a selective marker, however the strain must also carry Cas9 and gRNA expressing cassettes [4][11][12]. (4) I-SceI-assisted integration has a significantly increased efficiency, but this approach also requires the additional expression of the meganuclease I-SceI locus [13]. (5) Plas-mids carrying integrative cassettes excised by restriction with extended homology regions flanking the insert significantly improve integration efficiency [6][9][14]. We were inspired by this last strategy and decided to create a set of yeast integration cassettes that contain extended homology regions corresponding to a common selective marker locus, which eliminates the need to have a marker locus inside the cassette. An advantage of not having a selective marker locus inside the cassette reduces its size and potentially increases integration efficiency [8]. To reduce multiple integration time and to maximize available markers we constructed novel bidirectional promoter sets (Figure 1A).
–
The pDK series includes 24 plasmids which carry an integrative module for 4 common genetic markers (HIS3, URA3, ADE2, and TRP1). Constitutive (TEF1p, GPD1p), daughter specific (DSE4p) and inducible (CUP1p and bidirectional GAL1/10p) promoters flank multiple cloning sites. We also include 4 bidirectional promoters (GAL1p-GAL10p, TEF1p-GPD1p, TEF1p-CUP1p, and TEF1p-DSE4p) which allow multiple integrations (Figure 1, Table 1). PCR is carried out with short primers specific to a selectable marker in order to integrate the module (see Supplemental Table 1). The pDK set can be used for multi-color imaging, metabolic engineering, or any set of experiments that require stable genomic integration of one or more genes.
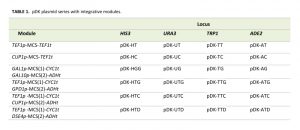 | TABLE 1. pDK plasmid series with integrative modules. |
RESULTS AND DISCUSSION
Vector overview
We designed 24 pDK plasmids for stable expression of multiple genes in the yeast S. cerevisiae (Figure 1, Table 1). Vectors can be used for stable integration into 4 common S. cerevisiae selective marker loci HIS3, URA3, ADE2, and TRP1. Plasmid variations include 6 promoters (Figure 1C): Constitutive TEF1p, inducible CUP1p, inducible bidirectional GAL1-10p in opposing orientation, constitutive bidirectional TEF1-GPD1p, constitutive-inducible bidirectional TEF1-CUP1p, constitutive-daughter-specific bidirectional TEF1-DSE4p (bidirectional promoter sets have two multiple cloning sites (MCS) in opposing orientation). The markers are split into two parts (see plasmid construction for details) and are flanking the region containing a promoter, MCS, and a terminator (Figure 1A). The fragment is amplified with primers specific to the marker (Supplemental Table 1). The orientation of the split markers allows integration of amplified region in a traditional homologous recombination driven way, resulting in doubling of the marker region (Figure 1B).
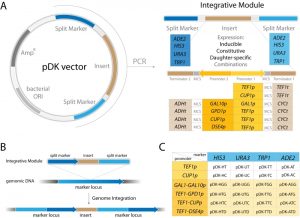 | FIGURE 1: pDK vector series overview. (A) pDK vector with an integrative module flanked by a split marker. The customized insert is flanked by constitutive, inducible, or daughter-specific promoters. PCR is required for genetic integration. (B) Integration overview: the module is transformed into S. cerevisiae and inserted into the chromosome resulting in a marker locus duplication. (C) pDK vector set: constitutive, inducible, and four bi-directional promoter plasmids available for integration into four markers. |
–
Integrative modules allow stable and efficient integration of multiple inserts
pDK vectors were examined for integration efficiency, correct integration, and stability of the insert. 24 integrative modules of pDK series were transformed into the W303 strain. pDK integrative modules have comparable integration efficiencies (Supplemental Figure 1A). Although double integration is possible we recommend sequential integration and using bidirectional promoters to expedite the work flow. We also compared the integration efficiency to other integration strategies: (1) pRS series – conventional linearized integrative plasmids [1], (2) EasyClone strategy – extended homology regions, excisable marker and restriction based integration [6], and (3) extended primers – 45bp homology region. In general, extended homology region based strategies provide higher efficiency of integration. pDK series integration is more effective than pRS series, and is comparable with EasyClone efficiency, which also has a reduced insert size but relies on restriction based integration.
–
To evaluate integration stability, a GFP reporter was cloned into the pDK series. The stability of the integration was tested by avoiding selection for more than 80 generations and then counting fluorescent cells (Supplemental Figure 1B). Modules are integrated with 95 – 100% accuracy into the marker region as verified by PCR (Supplemental Figure 1B). Marker based integration is advantageous because it provides accurate targeting compared to the CRISPR/Cas9 strategy [15]. We also tested the stability of multiple integrations, since many of them carry same promoters and/or terminators, which can potentially lead to a loop-out of the fragments. Single and multiple integrations provide reliable homogenous expression levels under non-selective conditions (Supplemental Figure 1B, C).
–
Application of pDK plasmids to multi-color imaging reveals order of organelle inheritance
As proof of concept for multi-color imaging we constructed peroxisomal, nuclear, and actin [16] markers using the pDK vector series and used the strain for 3D time-lapse microscopy (4D imaging) (Figure 2) [17]. Nuclear localization signal fused to a far-red fluorophore was used to visualize the nucleus. Peroxisome localization signal – tripeptide SKL was fused to the mCherry C-terminus to visualize peroxisomes. We also used LifeAct fused to GFP to visualize actin in living yeast [16]. The vacuolar marker VPH1 was endogenously tagged with mBFP or GFP. Peroxisomes are known to use actin cables for inheritance [18]. Inp2, an integral peroxisomal membrane protein, binds the Myo2 motor ensuring localization to the bud [19][20]. Most of the organelles in yeast use actin cables for bud trafficking during division [21]. To determine whether organelle inheritance proceeds in a parallel or a serial way we examined the inheritance of the vacuole, peroxisomes, and the nucleus (Figure 2, Supplemental movie “wtdivision”). The nucleus itself is inherited in a microtubule dependent manner, however the astral microtubules are delivered on actin to the bud [22]. Multi-color imaging revealed that the first instance of peroxisome inheritance happens in parallel with the vacuole in the first 15 min of cell division (Figure 2B). Deletion of INP2 abolishes peroxisome inheritance, which has no effect on the time of vacuole inheritance [19] (Figure 2C, Supplemental movie “inpdivision”). Interestingly, the nucleus is inherited prior to the end of cell division. The integrative vector series that we have developed allows for efficient integration of up to 8 markers, enabling us to image cellular compartments simultaneously.
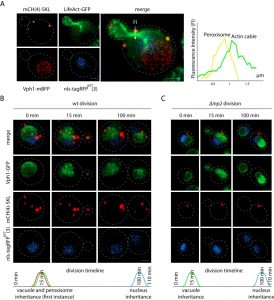 | FIGURE 2: Proof of concept - Organelle inheritance order during division. (A) In vivo 4-color imaging of peroxisome (SKL signal C-terminally fused to 4 mCherry (mCH) on pDK-UT), actin (LifeAct fused to GFP on pDK-AT), nucleus (SV40 nls signal fused to 3 far-red fluorophores on pDK-HT), and vacuole (VPH1 endogenously tagged with mBFP), the scale bar is 1 µm. The graph represents proximity of peroxisome to actin cable during division, Fluorescence Intensity (FI) was calculated along the x axes (arrow on the merge image) for red (peroxisome) and green (actin) channels. (B) The timeline of organelle inheritance in the wild type (wt) strain, scale bar is 1 µm, time points represent the average (n = 30) time of organelle inheritance from the start of division, strain carries SKL signal C-terminally fused to 4 mCherry (mCH) on pDK-UT module, SV40 nls signal fused to 3 far-red fluorophores on pDK-HT module, and VPH1 endogenously tagged with GFP. (C) The timeline of the organelle inheritance in the Δinp2 strain, scale bar is 1 µm. |
–
Application of pDK plasmids to multi-copy integration
As proof of concept of exquisitely controlled inducible expression of a gene using multiple integrations we inserted GFP tagged VHL under inducible CUP1 promoter in 4 marker loci. The von Hippel Landau tumor suppressor (VHL) has often been used as a model substrate to study protein aggregation in yeast [23]. Carefully controlling expression levels of integrated proteins is essential to many studies. In the case of VHL and similar proteins, increased protein concentration results in gradual protein self-association with subsequent decrease in the monomer fraction in vitro. This leads to the formation of protein inclusions or aggregates. To test if there is a correlation of misfolded protein concentration and inclusion formation in vivo we introduced 1-4 copies of GFP-VHL under CUP1p (Figure 3A, B) in yeast. We scored inclusion formation as a function of concentration and temperature. Aggregation strongly correlates with an increase in temperature, r(temperature) = 0.76 (p < 0.05), but has no significant correlation with VHL concentration, r(concentration) = 0.005 (p = 0.98). This could indicate a number of things. One interpretation of these data is that, regardless of the concentration, GFP-VHL forms a small percentage of overall protein content in inclusions (because of the abundance of misfolded and unstructured proteins in yeast). Another possibility is that without significant heat shock the quality control system is able to degrade a spectrum of misfolded VHL concentrations, but at higher temperatures it becomes uniformly inhibited. Additional experiments could make similar titrations of proteasome and chaperone activity. Together, these data are compelling as proof-of-concept for multi-gene integrations.
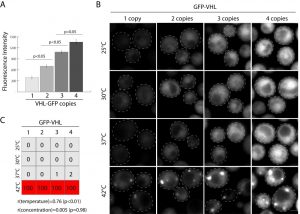 | FIGURE 3: Proof of concept - Concentration does not correlate with inclusion formation. (A) Gradual increase in the amount of GFP-VHL according to integrated copies, Fluorescence Intensity was quantified in 30 single cells in the population, the average and standard errors are represented on the graph. (B) Confocal images of cells carrying 4 copies of GFP-VHL on pDK-AC, TC, UC, HC modules subjected to the range of temperatures for 1h, scale bar 1 µm. (C) Quantification of inclusion forming cells in the population according to the change in the temperature and the concentration, the numbers represent % of the cells with inclusions (n = 300), correlation coefficient of aggregation and temperature (r(temperature)) and aggregation and concentration (r(concentration)) is provided under the diagram. |
–
Bidirectional promoter sets
To improve the time of strain construction, we built versatile bidirectional promoter sets, allowing integration of two inserts under different promoters at the same time. The promoters are positioned in the opposite orientation (Figure 1A). Previously described GAL1p/GAL10p and 3 novel promoters: TEF1p-GPD1p, TEF1p-CUP1p, and TEF1p-DSE4p were introduced into pDK series. To validate promoter sets we constructed GFP/mCherry reporters and visualized the cells during the log phase (Figure 4). Bidirectional promoters allow controlled inducible, semi-inducible, constitutive and daughter-specific expression (Figure 4A-D). Not only do they facilitate strain construction, bidirectional sets can also be used for cell sorting (daughter/mother cells), studying aging, or screening yeast genome collections [24][25].
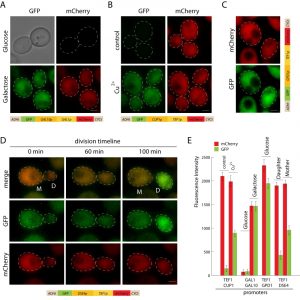 | FIGURE 4: Bidirectional promoter reporters. (A) Galactose inducible bidirectional promoter, cells carrying pDK-HGG-GFP-mCherry were induced with galactose or grown on glucose for 6 hours, scale bar 1 µm. (B) Constitutive-inducible promoter, cells carrying pDK-HTC-GFP-mCherry were grown with and without (control) copper2+ for 4 hours, scale bar 1 µm. (C) Constitutive bidirectional promoter, cells carrying pDK-HTG-GFP-mCherry were grown on glucose containing medium to middle log phase, scale bar 1 µm. (D) Daughter-specific-constitutive bidirectional promoter, cells carrying pDK-HTD-GFP-mCherry module were grown on glucose containing medium on the microscope, frames are shown (0 min, 60 min, 100 min). (E) Comparison of fluorescence intensity of bidirectional reporters, the graph shows the average of fluorescence intensity of 20 cells and standard error. |
–
In summary, the pDK vector series allows for efficient multiple integrations and thus is a useful tool for multi-color imaging, metabolic engineering, controlled expression of genes of interest, and stable yeast strain production. We therefore hope that pDK vectors will be a useful tool for the yeast community.
MATERIALS AND METHODS
Strains and media
We used standard conditions for culturing yeast and bacterial cells [26]. Yeast were grown in the selective medium (1.7 g/l yeast nitrogen base without amino acids and ammonium sulfate (Difco Laboratories), 5 g/l ammonium sulphate, 0.77 g/l complete, 2 g/l amino acids supplement powder mix [27], 20 g/l glucose, and 20 g/l agar for the solid medium) or rich medium (20 g/l Peptone, 10 g/l yeast extract, 20 g/l glucose, and 20 g/l agar for the solid medium). Galactose induction was performed on selective medium supplemented with 20 g/l galactose instead of glucose for 6 hours. CUP1p induction was performed by addition of 50 µM Cupric Sulphate to the selective medium and growing cells for 4 hours. Plasmids were constructed using Escherichia coli strain DH5α. Yeast strain W303-1B (MATα leu2-3,112 trp1-1 can1-100 ura3-1 ade2-1 his3-11,15) was used for the experiments.
 | TABLE 2. Strains used in this study. |
–
Plasmid construction
pUC19 plasmid [28] was used as a backbone for pDK vectors. Primers used in this study are summarized in supplemental tables: 1 – primers used for yeast integration, 2 – primers used for construction of plasmids. Markers loci (including promoters and terminators) were split in the following parts: HIS3 (after 286bp of the ORF), URA3 (after 420 bp of the ORF), TRP1 (after 224 bp of the ORF), ADE2 (after 462 bp of the ORF). Split fragments were amplified with: HIS3F1/R1 and HIS3F2/R2, URA3F1/R1 and URA3F2/R2, TRP1F1/R1 and TRP1F2/R2, ADE2F1/R1 and ADE2F2/R2, and cloned into pUC19’s PciI/HindIII and PfoI/EcoRI, EcoRI/PfoI and PciI/HindIII, HindIII/PciI and PfoI/EcoRI, HindIII/PciI and PfoI/EcoRI respectively, resulting in the plasmids pDK-HH, pDK-UU, pDK-TT, and pDK-AA. Promoters and terminators (TEF1p, CUP1p, TEF1t) were amplified with: TEF1PF/R, CUP1PF/R, TEF1TF/R primers and cloned into pUC19’s NdeI/EcoRI, NdeI/EcoRI, and SalI/HindIII sites, respectively, resulting in pUC19-TEF1p–TEF1t and pUC19-CUP1p–TEF1t plasmids. TEF1p–TEF1t, CUP1p–TEF1t and GAL1/10p–ADH1/CYC1t modules were amplified from previously constructed pUC19-TEF1p–TEF1t and pUC19-CUP1p–TEF1t, and pESC-URA (Agilent) vectors using the primers: CUPTEFHHF, CUPTEFUUF, CUPTEFTTF, CUPTEFAAF and CUPTEFR for TEF1p–TEF1 and, CUP1p–TEF1t modules, and GALHHF, GALUUF, GALTTF, GALAAF and GALR for GAL1/10p–ADH1/CYC1t module, and cloned using Gibson Assembly mix (NEB) into pDK-HH, pDK-UU, pDK-TT, and pDK-AA vectors linearized with EcoRI/SalI restriction enzymes resulting in 12 pDK plasmids. Plasmid names contain the first letter of the marker followed by the first letter/s of the promoter/s (Table 1). TEF1 promoter was amplified with TEFbF/R primers and cloned into pESC-URA AgeI/BamHI sites resulting in the pESC-TEFp plasmid. CUP1, DSE4, and GPD1 promoters were amplified with CUPbF/R, DSEbF/R, and GPDbF/R primers and cloned into the pESC-TEFp vector into AgeI/EcoRI sites resulting in pESC-TG, TC and TD plasmids carrying bidirectional promoters. Bidirectional promoters were subcloned using BamHI/EcoRI into pDK-HG/UG/TG/AG, resulting in 12 bidirectional promoter sets (Table 1).
–
The nuclear cellular marker plasmid was constructed by cloning the SV40 nuclear localization signal fused to (tagRFP657)4 [29] into pDK-HT vector EcoRI/SmaI sites. The peroxisome marker was constructed by cloning the (mCherry)4-SKL sequence into pDK-UT plasmid. Actin was visualized via the LifeAct fragment fused to GFP [16], LifeAct-GFP was cloned into the pDK-AT vector. VHL plasmids were constructed by cloning the GFP-VHL sequence into pDK-HC, pDK-UC, pDK-AC, and pDK-TC vectors. pCfB2513 was a gift from Irina Borodina (Addgene plasmid # 67543).
–
For endogenous tagging we modified the pKT127 [30] plasmid by inserting the mBFP sequence or GFP sequence into PacI/AscI sites and HPH marker sequence into BglII/PmeI sites. GFP reporters were constructing by cloning GFP into SacI/XmaI sites of pDK-HT/HC, and BamHI/XmaI sites of pDK-HGG with primers GFPxR, GFPbF, GFPsF and subcloning the fragment containing the marker across different marker plasmids and bidirectional promoter ones. mCherry was amplified with primers CHeF and CHnR and cloned into the EcoI/SpeI sites of pDK-HGG-GFP, and subcloned to pDK-HTC-GFP, pDK-HTG-GFP, and pDK-HTD-GFP.
–
Strain construction
We introduced the INP2 deletion using a PCR based deletion strategy [31] and primers delINPF/R. Gene deletions were verified by PCR. Strains with integrative modules were constructed by transforming yeast with a PCR fragment obtained from a corresponding plasmid with a set of primers listed in Table 1. For comparison experiments pRS plasmids were linearized in the marker locus prior to integration, pRS303 and pRS306 with PstI restriction enzyme, and pRS304 with PmlI enzyme.
–
VPH1 was tagged using modified pKT127 plasmid and eVPHF/R primers.
–
Protocol for yeast integration
The fragment for genomic integration is generated via PCR with primers listed in Table 2 using the following parameters (95°C- 5’, [95°C- 30’’, 62°C- 30’’ (increment 0.8°C per cycle), 72°C- X min (X = length of the fragment in kb)]25 cycles, 72°C- 5’), and high fidelity polymerase generating blunt-end products, e.g. KAPA HiFi DNA Polymerase (KAPA Biosystems). A PCR protocol with fixed primer binding time can also be used. Up to 2 μg of PCR product is transformed using LiAc/PEG transformation [32] with modifications – 50 μl of DMSO is added prior to heat shock, heat shock time is reduced to 15 min at 42°C. 20 random clones carrying integrative modules were verified by PCR.
–
Stability of integration
Yeast strains were grown in rich medium in three replicates. The culture was diluted 1/100 every 24 hours for 10 days. The culture was analyzed by reporter fluorescence on the first and tenth day.
–
Confocal Microscopy
For imaging yeast cells were grown to mid-log phase and seeded on concanavalin A (Sigma) coated 4-well microscope plates (IBIDI). For induction galactose rich media (division experiments, Figure 2) or minimal media with 50 µm Cu2+ (Figure 3) were used. Copper induced cells were grown in 25°C to mid-log phase and then incubated for 1h at indicated temperatures (30, 37, 42°C). Confocal 3D images and movies were acquired using a dual point-scanning Nikon A1R-si microscope equipped with a PInano Piezo stage (MCL), using a 60 x PlanApo VC oil objective NA 1.40. Movies were acquired in resonant-scanning mode. Image processing was performed using NIS-Elements software.
–
Statistics
The experiments were repeated at least 3 times. Multiple correlation coefficients were calculated for 3 variables (aggregation, temperature, concentration), with subsequent regression analysis to determine p-values. Standard comparisons were performed using t-test.
References
- R.S. Sikorski, and P. Hieter, "A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae.", Genetics, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2659436
- F. Fang, K. Salmon, M.W.Y. Shen, K.A. Aeling, E. Ito, B. Irwin, U.P.C. Tran, G.W. Hatfield, N.A. Da Silva, and S. Sandmeyer, "A vector set for systematic metabolic engineering in Saccharomyces cerevisiae", Yeast, vol. 28, pp. 123-136, 2010. http://dx.doi.org/10.1002/yea.1824
- S. Alberti, A.D. Gitler, and S. Lindquist, "A suite of Gateway® cloning vectors for high‐throughput genetic analysis in Saccharomyces cerevisiae", Yeast, vol. 24, pp. 913-919, 2007. http://dx.doi.org/10.1002/yea.1502
- C. Ronda, J. Maury, T. Jakočiu̅nas, S.A. Baallal Jacobsen, S.M. Germann, S.J. Harrison, I. Borodina, J.D. Keasling, M.K. Jensen, and A.T. Nielsen, "CrEdit: CRISPR mediated multi-loci gene integration in Saccharomyces cerevisiae", Microbial Cell Factories, vol. 14, 2015. http://dx.doi.org/10.1186/s12934-015-0288-3
- N. Agmon, L.A. Mitchell, Y. Cai, S. Ikushima, J. Chuang, A. Zheng, W. Choi, J.A. Martin, K. Caravelli, G. Stracquadanio, and J.D. Boeke, "Yeast Golden Gate (yGG) for the Efficient Assembly of S. cerevisiae Transcription Units", ACS Synthetic Biology, vol. 4, pp. 853-859, 2015. http://dx.doi.org/10.1021/sb500372z
- V. Stovicek, G.M. Borja, J. Forster, and I. Borodina, "EasyClone 2.0: expanded toolkit of integrative vectors for stable gene expression in industrial Saccharomyces cerevisiae strains", Journal of Industrial Microbiology and Biotechnology, vol. 42, pp. 1519-1531, 2015. http://dx.doi.org/10.1007/s10295-015-1684-8
- F.C.B. Leite, R.S.G. dos Anjos, A.C.M. Basilio, G.F.C. Leal, D.A. Simões, and M.A. de Morais, "Construction of integrative plasmids suitable for genetic modification of industrial strains of Saccharomyces cerevisiae", Plasmid, vol. 69, pp. 114-117, 2013. http://dx.doi.org/10.1016/j.plasmid.2012.09.004
- S.H. Kung, A.C. Retchless, J.Y. Kwan, and R.P.P. Almeida, "Effects of DNA Size on Transformation and Recombination Efficiencies in Xylella fastidiosa", Applied and Environmental Microbiology, vol. 79, pp. 1712-1717, 2013. http://dx.doi.org/10.1128/Aem.03525-12
- N.B. Jensen, T. Strucko, K.R. Kildegaard, F. David, J. Maury, U.H. Mortensen, J. Forster, J. Nielsen, and I. Borodina, "EasyClone: method for iterative chromosomal integration of multiple genes Saccharomyces cerevisiae", FEMS Yeast Research, vol. 14, pp. 238-248, 2014. http://dx.doi.org/10.1111/1567-1364.12118
- M. Jahn, C. Vorpahl, T. Hübschmann, H. Harms, and S. Müller, "Copy number variability of expression plasmids determined by cell sorting and Droplet Digital PCR", Microbial Cell Factories, vol. 15, 2016. http://dx.doi.org/10.1186/s12934-016-0610-8
- A. Horwitz, J. Walter, M. Schubert, S. Kung, K. Hawkins, D. Platt, A. Hernday, T. Mahatdejkul-Meadows, W. Szeto, S. Chandran, and J. Newman, "Efficient Multiplexed Integration of Synergistic Alleles and Metabolic Pathways in Yeasts via CRISPR-Cas", Cell Systems, vol. 1, pp. 88-96, 2015. http://dx.doi.org/10.1016/j.cels.2015.02.001
- S. Shi, Y. Liang, M.M. Zhang, E.L. Ang, and H. Zhao, "A highly efficient single-step, markerless strategy for multi-copy chromosomal integration of large biochemical pathways in Saccharomyces cerevisiae", Metabolic Engineering, vol. 33, pp. 19-27, 2016. http://dx.doi.org/10.1016/j.ymben.2015.10.011
- N.G. Kuijpers, S. Chroumpi, T. Vos, D. Solis-Escalante, L. Bosman, J.T. Pronk, J. Daran, and P. Daran-Lapujade, "One-step assembly and targeted integration of multigene constructs assisted by the I-SceI meganuclease inSaccharomyces cerevisiae", FEMS Yeast Research, vol. 13, pp. 769-781, 2013. http://dx.doi.org/10.1111/1567-1364.12087
- C. Taxis, and M. Knop, "System of Centromeric, Episomal, and Integrative Vectors Based on Drug Resistance Markers for SacCharomyces Cerevisiae", BioTechniques, vol. 40, pp. 73-78, 2006. http://dx.doi.org/10.2144/000112040
- S.W. Cho, S. Kim, Y. Kim, J. Kweon, H.S. Kim, S. Bae, and J. Kim, "Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases", Genome Research, vol. 24, pp. 132-141, 2013. http://dx.doi.org/10.1101/gr.162339.113
- J. Riedl, A.H. Crevenna, K. Kessenbrock, J.H. Yu, D. Neukirchen, M. Bista, F. Bradke, D. Jenne, T.A. Holak, Z. Werb, M. Sixt, and R. Wedlich-Soldner, "Lifeact: a versatile marker to visualize F-actin", Nature Methods, vol. 5, pp. 605-607, 2008. http://dx.doi.org/10.1038/nmeth.1220
- R. Spokoini, M. Shamir, A. Keness, and D. Kaganovich, "4D Imaging of Protein Aggregation in Live Cells", Journal of Visualized Experiments, 2013. http://dx.doi.org/10.3791/50083
- D. Hoepfner, M. van den Berg, P. Philippsen, H.F. Tabak, and E.H. Hettema, "A role for Vps1p, actin, and the Myo2p motor in peroxisome abundance and inheritance in Saccharomyces cerevisiae ", The Journal of Cell Biology, vol. 155, pp. 979-990, 2001. http://dx.doi.org/10.1083/jcb.200107028
- A. Fagarasanu, M. Fagarasanu, G.A. Eitzen, J.D. Aitchison, and R.A. Rachubinski, "The Peroxisomal Membrane Protein Inp2p Is the Peroxisome-Specific Receptor for the Myosin V Motor Myo2p of Saccharomyces cerevisiae", Developmental Cell, vol. 10, pp. 587-600, 2006. http://dx.doi.org/10.1016/j.devcel.2006.04.012
- R. Saraya, M.N. Cepińska, J.A. Kiel, M. Veenhuis, and I.J.V. der Klei, "A conserved function for Inp2 in peroxisome inheritance", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1803, pp. 617-622, 2010. http://dx.doi.org/10.1016/j.bbamcr.2010.02.001
- B. Knoblach, and R.A. Rachubinski, "Sharing the cell's bounty – organelle inheritance in yeast", Journal of Cell Science, 2015. http://dx.doi.org/10.1242/jcs.151423
- D.L. Beach, J. Thibodeaux, P. Maddox, E. Yeh, and K. Bloom, "The role of the proteins Kar9 and Myo2 in orienting the mitotic spindle of budding yeast", Current Biology, vol. 10, pp. 1497-1506, 2000. http://dx.doi.org/10.1016/S0960-9822(00)00837-X
- K. Brock, A. Abraham, T. Amen, D. Kaganovich, and J. England, "Structural Basis for Modulation of Quality Control Fate in a Marginally Stable Protein", Structure, vol. 23, pp. 1169-1178, 2015. http://dx.doi.org/10.1016/j.str.2015.04.015
- A. Li, Z. Liu, Q. Li, L. Yu, D. Wang, and X. Deng, "Construction and characterization of bidirectional expression vectors inSaccharomyces cerevisiae", FEMS Yeast Research, vol. 8, pp. 6-9, 2008. http://dx.doi.org/10.1111/j.1567-1364.2007.00335.x
- B. Afonso, P.A. Silver, and C.M. Ajo-Franklin, "A synthetic circuit for selectively arresting daughter cells to create aging populations", Nucleic Acids Research, vol. 38, pp. 2727-2735, 2010. http://dx.doi.org/10.1093/nar/gkq075
- F. Sherman, "Getting started with yeast.", Methods in enzymology, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12073320
- A. Baryshnikova, M. Costanzo, S. Dixon, F.J. Vizeacoumar, C.L. Myers, B. Andrews, and C. Boone, "Synthetic Genetic Array (SGA) Analysis in Saccharomyces cerevisiae and Schizosaccharomyces pombe", Methods in Enzymology, pp. 145-179, 2010. http://dx.doi.org/10.1016/s0076-6879(10)70007-0
- C. Yanisch-Perron, J. Vieira, and J. Messing, "Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mpl8 and pUC19 vectors", Gene, vol. 33, pp. 103-119, 1985. http://dx.doi.org/10.1016/0378-1119(85)90120-9
- K.S. Morozova, K.D. Piatkevich, T.J. Gould, J. Zhang, J. Bewersdorf, and V. Verkhusha, "Far-Red Fluorescent Protein Excitable with Red Lasers for Flow Cytometry and Superresolution STED Nanoscopy", Biophysical Journal, vol. 99, pp. L13-L15, 2010. http://dx.doi.org/10.1016/j.bpj.2010.04.025
- M.A. Sheff, and K.S. Thorn, "Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae", Yeast, vol. 21, pp. 661-670, 2004. http://dx.doi.org/10.1002/yea.1130
- A. Baudin, O. Ozier-Kalogeropoulos, A. Denouel, F. Lacroute, and C. Cullin, "A simple and efficient method for direct gene deletion inSaccharomyces cerevisiae", Nucleic Acids Research, vol. 21, pp. 3329-3330, 1993. http://dx.doi.org/10.1093/nar/21.14.3329
- R.D. Gietz, R.H. Schiestl, A.R. Willems, and R.A. Woods, "Studies on the transformation of intact yeast cells by the LiAc/SS‐DNA/PEG procedure", Yeast, vol. 11, pp. 355-360, 1995. http://dx.doi.org/10.1002/yea.320110408
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We thank members of the Jerusalem Brain Community for their support. This work was supported by the European Research Council under the European Union's Seventh Framework Programme (FP/2007-2013)/ERC-StG2013 337713 DarkSide starting grant, as well as an Israel Science Foundation Grant ISF 843/11; a German Israel Foundation Grant GIFI-1201-242.13/2012; a Niedersachsen-Israel Research Program grant, an Abisch-Frenkel Foundation grant, and a joint Israel-Italy cooperation grant from the Israeli Ministry of Science, Technology, and Space. TA was funded by a Jerusalem Brain Community Doctoral Fellowship and by the Alexander Grass Center for Bioengineering.
COPYRIGHT
© 2017

Integrative modules for efficient genome engineering in yeast by Amen and Kaganovich is licensed under a Creative Commons Attribution 4.0 International License.








