
Research Articles:
Microbial Cell, Vol. 1, No. 1, pp. 9 - 20; doi: 10.15698/mic2014.01.120
Tracking autophagy during proliferation and differentiation of Trypanosoma brucei
1 Wellcome Trust Centre for Molecular Parasitology, Institute of Infection, Immunity and Inflammation, College of Medical, Veterinary and Life Sciences, University of Glasgow, Glasgow, G12 8TA, UK.
2 Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, Glasgow, G4 0RE, UK.
Keywords: trypanosome, protozoan, autophagy, differentiation, cell death.
Received originally: 28/10/2013 Received in revised form: 06/12/2013
Accepted: 13/12/2013
Published: 06/01/2014
Correspondence:
Jeremy C. Mottram, 120 University Place; Glasgow G12 8TA, UK. jeremy.mottram@glasgow.ac.uk
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: William R. Proto, Nathaniel G. Jones, Graham H. Coombs, and Jeremy C. Mottram (2014). Tracking autophagy during proliferation and differentiation of Trypanosoma brucei. Microbial Cell 1(1): 9-20. doi: 10.15698/mic2014.01.120
Abstract
Autophagy is a lysosome-dependent degradation mechanism that sequesters target cargo into autophagosomal vesicles. The Trypanosoma brucei genome contains apparent orthologues of several autophagy-related proteins including an ATG8 family. These ubiquitin-like proteins are required for autophagosome membrane formation, but our studies show that ATG8.3 is atypical. To investigate the function of other ATG proteins, RNAi compatible T. brucei were modified to function as autophagy reporter lines by expressing only either YFP-ATG8.1 or YFP-ATG8.2. In the insect procyclic lifecycle stage, independent RNAi down-regulation of ATG3 or ATG7 generated autophagy-defective mutants and confirmed a pro-survival role for autophagy in the procyclic form nutrient starvation response. Similarly, RNAi depletion of ATG5 or ATG7 in the bloodstream form disrupted autophagy, but did not impede proliferation. Further characterisation showed bloodstream form autophagy mutants retain the capacity to undergo the complex cellular remodelling that occurs during differentiation to the procyclic form and are equally susceptible to dihydroxyacetone-induced cell death as wild type parasites, not supporting a role for autophagy in this cell death mechanism. The RNAi reporter system developed, which also identified TOR1 as a negative regulator controlling YFP-ATG8.2 but not YFP-ATG8.1 autophagosome formation, will enable further targeted analysis of the mechanisms and function of autophagy in the medically relevant bloodstream form of T. brucei.
INTRODUCTION
Autophagy is a primary intracellular mechanism for the degradation of long lived proteins and organelles. It describes multiple distinct pathways that all utilise the lysosome (or vacuole) to degrade and recycle unwanted cellular components. Of these pathways, the major form is macroautophagy (hereafter referred to as autophagy), which is characterised by the engulfment of cargo destined for the lysosome into a double membrane bound vesicle called an autophagosome [1]. Autophagy was once regarded solely as a stress response mechanism enabling cells to withstand nutrient starvation; however it is now clear that it participates in wide ranging cellular processes influencing aspects of growth, development, differentiation and immunity [2]. The dramatic expansion in the known functions of autophagy stems largely from the improved definition of the pathway’s molecular mechanisms achieved through extensive study of model systems such as yeast. This has led to the identification of multiple autophagy related genes (ATGs) that control and execute a complex process broadly summarised by the following principle stages – induction, autophagosome formation and degradation.
–
Autophagy is subject to tight regulation to ensure that it functions optimally. The signalling pathways that control autophagy induction are organism specific, but typically function through the protein kinase, Atg1, and its interacting partners [3]. Post induction, the isolation membrane expands to engulf and eventually surround the cargo in a mature autophagosome [4]. This process requires the coordinated recruitment of multiple factors, including components of two ubiquitin-like conjugation systems (Atg8-phosphatidylethanolamine (PE) and Atg12-Atg5) that participate in membrane elongation and autophagosome completion [5],[6],[7]. Atg12 is a small ubiquitin-like protein (UBL) that covalently links to Atg5 in a process requiring Atg7 and Atg10. Post conjugation, Atg16 associates with Atg5, stimulating homo-multimer formation, which facilitates membrane binding and Atg8 recruitment [8],[9]. During membrane expansion the UBL Atg8 is uniquely conjugated to PE, an abundant membrane phospholipid [6]. This process requires the concerted action of Atg4, Atg7 and Atg3, and produces a lipidated protein that tightly associates with membrane. The Atg8-PE that decorates the external surface of the mature autophagosome is removed by the proteolytic action of Atg4, whereas internal Atg8-PE remains associated [10]. To recycle the autophagosome contents, the external autophagosome membrane fuses with the lysosome and its cargo, still bound by the inner membrane, is then degraded.
–
In silico screening of genome databases for ATG genes has indicated that autophagy is widely conserved amongst eukaryotes, and this has been confirmed experimentally with some species including evolutionary divergent parasitic protozoa, such as the kinetoplastids and apicomplexans [11],[12],[13],[14]. Trypanosoma brucei is a parasitic protozoon, transmitted by the tsetse fly, which causes Human and Animal African Trypanosomiasis (HAT and AAT). During its life cycle, the parasite progresses through a number of developmental stages within the insect and mammalian hosts. Long slender bloodstream form (BSF) trypomastigotes replicate within the mammalian host, whilst procyclic form (PCF) trypomastigotes replicate within the tsetse mid gut. The short stumpy bloodstream trypomastigote is an intermediate transition life cycle stage that is transmission competent. The T. brucei genome contains identifiable genes encoding for approximately half of the yeast Atg proteins, including components of both the ATG8 and ATG12 conjugation pathways [11],[12],[13].
–
Investigation of autophagy in protozoan parasites has revealed that autophagy is central to fundamental processes such as cellular differentiation, virulence and the starvation response (reviewed in [15]). In addition to these pro-survival functions, autophagy has also been linked to a form of regulated cell death, known as autophagic cell death, in some protozoan parasites [16],[17]. These findings are controversial, however, and detailed molecular analyses are required to identify whether activation of autophagy has occurred to orchestrate cell death or is merely a response to cell stress coincident with cell death [18]. Accordingly, it is important to elucidate whether or not autophagy is an active death mechanism; for instance, by using targeted genetic or chemical manipulation of the autophagy pathway and determining if this delays or abolishes the cell death phenotype under investigation.
–
Eukaryotic Target of Rapamycin (TOR) regulates diverse cellular events, most notably, in the context of this study the induction of autophagy. TOR functions through two multi-component complexes to control distinct aspects of cellular growth; TOR complex 1 (TORC1), which regulates temporal cell growth and TOR complex 2 (TORC2), which controls spatial cell growth [19]. Rapamycin is a macrolide widely used to induce autophagy and the compound’s selective inhibition of TORC1 suggests this is the complex responsible for autophagy control [20],[21]. T. brucei has both TORC1 and TORC2 complexes, but it has been reported that only TORC2 is sensitive to rapamycin and thus that rapamycin is not suitable for inducing autophagy in T. brucei [22].
–
In order to resolve some of these confounding issues we attempted a comprehensive analysis of T. brucei autophagy, focusing on both mechanism and physiological function. Confirming T. brucei ATG8.1 and ATG8.2 as bone fide ATG8-like proteins informed the subsequent development of transgenic T. brucei that combined autophagy-specific fluorescent reporter systems with RNAi silencing technology. Through RNAi targeting of genes predicted to function in the T. brucei autophagy pathway we were able to generate loss of function autophagy mutants. In PCF this identified a role for autophagy facilitating survival during nutrient starvation, but not in executing specific autophagic cell death events. In contrast, disruption of BSF autophagy did not lead to detectable phenotypes in cellular proliferation or differentiation.
RESULTS
Autophagy in procyclic form T. brucei
T. brucei has 3 ATG8-like genes, ATG8.1, ATG8.2 and ATG8.3 (TritrypDB gene ID: Tb927.7.5900, Tb927.7.5910 and Tb927.7.3320 respectively). Structural analysis of ATG8.2 showed the protein contains a classical ubiquitin-like fold and displays significant structural homology to bone fide ATG8s, including conservation of key functional residues required for Atg4 interaction and lipidation as identified in yeast [23]. The T. brucei ATG8.1 and ATG8.2 proteins share 82% amino acid identity (the only divergence occurring at their N-termini (Fig. S1)) and are both incorporated into autophagosomes in response to nutrient depletion of cultured procyclic form (PCF) T. brucei [16]. However the role of ATG8.3 remains unclear, as despite good sequence identity to ATG8.1 and ATG8.2, a large insertion is predicted to interfere with the ubiquitin fold prohibiting interaction with ATG4 (Fig. S1)[23].
–
To investigate the 3 T. brucei ATG8-like genes we engineered PCF cell lines to conditionally express the proteins as N-terminal yellow fluorescent protein (YFP) fusions under the control of a tetracycline (tet) inducible promoter. The tet-induced expression of each fusion protein was verified by western blotting using anti-ATG8.1, an affinity purified polyclonal antibody raised against recombinant T. brucei ATG8.1 and anti-GFP, showing that the proteins were expressed at their predicted molecular masses (Fig. 1A). Only YFP-ATG8.1 and YFP-ATG8.2 were recognised by anti-ATG8.1, whilst YFP-ATG8.3 expression was only detected with anti-GFP antibody. While the anti-ATG8.1 confirmed that endogenous ATG8 expression occurred in both BSF and PCF wild type cell lines (Fig.1B), the similarity in the ATG8 predicted molecular masses (ATG8.1, 13.3 kDa; ATG8.2, 13.8 kDa; ATG8.3, 15.3 kDa), and the selective cross reactivity of anti-ATG8.1, prohibited identification of the endogenous ATG8 isoforms. Anti-ATG8.1 antibody also detected an unknown protein of approximately 40 kDa in both lifecycle stages (Fig. 1B).
–
Having established tight conditional expression of the ATG8 fusion proteins fluorescent microscopy was used to determine whether any characteristic ATG8 protein properties were evident. During standard nutrient rich culture YFP-ATG8.1 and YFP-ATG8.2 were predominantly distributed throughout the cytoplasm with punctate structures observed in a minority of cells. However, subjecting the tet induced cells to nutrient starvation, a widely used inducer of autophagy, significantly increased the number of observed puncta; the mean number of YFP-ATG8.1 puncta per cell increased from 0.22 (±0.03) to 1.30 (±0.25) and YFP-ATG8.2 puncta per cell increased from 0.13 (±0.07) to 0.87 (±0.08) (Figs 1C and 1D). These findings are in good agreement with data published previously showing autophagosome formation in PCF trypanosomes [16]. In contrast to the study by Li et al. [16] glucose supplementation of the starvation media was not required for puncta formation.
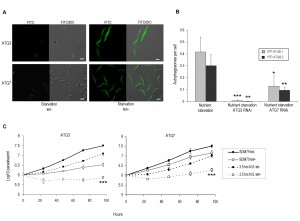
 | FIGURE 1: Conditional expression of YFP-ATG8 proteins in procyclic form T. brucei. (A) Expression of individual YFP-ATG8 fusion proteins was induced by growing cells for 24 h in media containing tetracycline (tet). 1 x 106 cells were loaded per lane and immuno-blotted with anti-ATG8.1 or anti-GFP antibody. Anti-EF1α was used to demonstrate equal loading. (B) Endogenous ATG8 expression. 5 x106 wild type BSF and PCF cells load-ed per lane, were probed with anti-ATG8.1. Anti-EF1α was used to demonstrate equal loading. (C) Fluorescent microscopy was used to monitor the presence of autophagosomes in PCF cells conditionally expressing YFP fusion proteins following growth in SDM79 media, starvation in PBS for 2.5 h or 2.5 h nutrient starvation in PBS supplemented with 10 µM wortmannin. The mean number of autophagosomes per cell was determined by counting >200 cells with data displayed as a mean of three replicate experiments from three inde-pendent clones. Error bars represent standard deviation and asterisks indicate where data differed significantly from the mean of the non-starved controls *p<0.05, ***p<0.001. (D) Representative images of cells assessed as for C. Left hand images FITC filter set, right hand images DIC FITC merge. Scale bar 5 µm. |
YFP-ATG8.3 was markedly different to YFP-ATG8.1 and YFP-ATG8.2, with comparatively low levels of puncta formation observed (Figs 1C and 1D). During standard culture conditions the mean puncta per cell was 0.02 (± 0.01), more than 10- and 5-fold lower than YFP-ATG8.1 and YFP-ATG8.2, respectively. Nutrient starvation did increase the observed puncta per cell to 0.04 (±0.03), however the magnitude of change was far lower than for YFP-ATG8.1 and YFP-ATG8.2. To provide more evidence for a link between puncta formation and the autophagy pathway, the effect of supplementing the nutrient starvation buffer with wortmannin, a PI3 kinase inhibitor and established autophagy inhibitor [16], was monitored (Figs 1C and 1D). As expected this reduced the appearance of puncta; effectively abolishing YFP-ATG8.1 and YFP-ATG8.3 puncta formation and significantly decreasing YFP-ATG8.2 puncta. These findings, in combination with those of Li et al. [16], provide convincing evidence that ATG8.1 and ATG8.2 are incorporated into autophagosomes and are therefore true ATG8-like proteins.
–
RNAi ablation of autophagy in procyclic form T. brucei
To provide functional insights into T. brucei autophagy the YFP-ATG8 expression constructs were modified to operate in conjunction with RNAi compatible T. brucei cell lines [24],[25]. To simplify the T. brucei autophagy reporter system, a plasmid designed for constitutive ectopic expression in T. brucei [26] was generated by switching the drug resistance to BSD and then inserting the YFP-ATG8.1 and –ATG8.2 ORFs into the expression site. The new constructs, were individually transfected into PCF 29-13 cells [24] already containing 2T7ti RNAi constructs [27] targeting the T. brucei orthologues of either ATG3 (Tb927.2.1890) or ATG7 (Tb927.10.11180). These genes were chosen as initial proof of concept targets based on presumed essentiality to the autophagy pathway and high confidence bioinformatic predictions indicating single copy orthologues in the T. brucei genome [11],[13],[15].
–
Microscopic analysis of the PCF cells confirmed that the constitutively expressed YFP-ATG8.1 and YFP-ATG8.2 fusion proteins were functional and formed autophagosomes during nutrient starvation (Figs 2A and 2B). RNAi depletion of both ATG3 and ATG7 orthologues for 72 h significantly decreased the number of autophagosomes observed per cell following starvation (Figs 2A and 2B). Although cells depleted of ATG3 were almost entirely devoid of puncta after starvation, the effect of ATG7 RNAi was less severe. This difference could be linked to RNAi penetration, with real time PCR confirming post RNAi depletion of ATG7 transcript occurs to a lesser extent than ATG3 (Fig. S2). However, it could also reflect varying half-lives of the target proteins, their different roles in the autophagy pathway or the presence of additional, and as yet unknown, ATG7 orthologues. Nevertheless, these findings provided additional experimental evidence supporting the identification of the T. brucei ATG3 and ATG7 orthologues [13],[16] and created PCF autophagy mutants for detailed loss of function analysis.
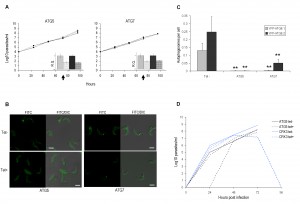
 | FIGURE 2: Procyclic form ATG3 and ATG7 RNAi mutants. Constitutive expression of YFP-ATG8.1 and YFP-ATG8.2 in PCF was visualised by fluorescent microscopy following RNAi of either ATG3 or ATG7 for 72 h before nutrient starvation in PBS for 2.5 h. (A) Representative images, left hand images FITC filter set, right hand images DIC FITC merge. Scale bar 5 µm. (B) The mean number of autophagosomes per cell was determined by counting >200 cells with data displayed as a mean of three replicate experiments. Error bars represent standard deviation and asterisks indicate where data differed significantly from the means of uninduced controls *p<0.05, ** p<0.01, ***p<0.001. (C) ATG3 and ATG7 RNAi was induced for 72 h in cells expressing YFP-ATG8.1 before nutrient starvation for 3.5 h in PBS (dashed lines) or control treatment in SDM79 media (solid lines). Cells were re-seeded at 0 h in SDM79 + tet (empty boxes grey line, tet+) and SDM79 –tet (filled boxes black line, tet-) and growth was determined daily for 96 h. Data represent mean of three replicate experiments. Error bars show standard deviation and asterisks indicate where data differed significantly from the means of tet induced starved and non-starved lines at the 96 h time point ***p<0.001. |
Independent induction of ATG3 or ATG7 RNAi impaired cell growth of PCF cultured in normal media (Fig. 2C). The expression of YFP-ATG8.1 or YFP-ATG8.2 did not appear to influence the RNAi growth phenotypes, as RNAi in the PCF 29-13 cells produced similar results (data not shown). In T. brucei PCF RNAi autophagy mutants have previously been used to implicate the autophagy machinery in an active cell death process during prolonged in vitro PCF nutrient deprivation [16]. However, in this study disruption of PCF autophagy by 72 h RNAi of ATG3 or ATG7, significantly diminished the capacity of the parasites to withstand and recover from acute nutrient starvation (Fig. 2C).
–
RNAi ablation of autophagy in bloodstream form T. brucei
Having effectively combined the YFP-ATG8 reporters with RNAi in PCFs we extended our analyses to the BSF of the parasite. The constitutive expression constructs for YFP-ATG8.1 and YFP-ATG8.2 were used to create RNAi autophagy reporter lines in the 2T1 BSF cell line [25]. The resulting parental 2T1 cell lines individually expressing YFP-ATG8.1 or YFP-ATG8.2, were therefore compatible with the stem loop RNAi vector pTL that had been specifically designed to facilitate rapid construct generation [28]. Initially, RNAi constructs were designed to target the T. brucei ATG5 and ATG7 orthologues. RNAi depletion of either ATG5 or ATG7 did not impact cell growth over 96 h (Fig. 3A), and therefore 72 h post induction was validated as an appropriate time point for autophagosome analysis with RNAi specificity confirmed by quantitative RT-PCR (Fig. 3A). As predicted, RNAi depletion of both ATG5 and ATG7 significantly decreased the number of autophagosomes observed (Fig. 3B and 3C). However, RNAi of ATG5 was the most effective at inhibiting puncta formation and was therefore selected for the subsequent loss of function analyses (Figs 3D, 4 and 5). To determine whether autophagy might contribute to parasite growth or survival in a more stringent in vivo environment, mice were infected with YFP-ATG8.2 ATG5 RNAi parasites and RNAi was induced 24 h later by supplementing a subset of the mice’s drinking water with doxycycline. This protocol allows clearance of parasites from the blood within 48 h for essential genes, such as the kinase CRK3 (Figs 3D and S3) [28]. However, no difference in parasitaemia was observed between control and induced ATG5 RNAi cell lines (Fig. 3D), suggesting that under the conditions tested autophagy was dispensable for the short term growth of monomorphic 2T1 BSF.
 | FIGURE 3: RNAi of autophagy genes in bloodstream form. (A) Growth analysis of BSF RNAi mutants in vitro. RNAi of ATG5 and ATG7 genes in BSF 2T1 constitutively expressing YFP-ATG8.1 (diamonds) and YFP-ATG8.2 (squares). RNAi induced with tetracycline (filled symbols) and growth compared to controls (empty symbols) for 96 h. Cells were re-seeded to 1 x 104 ml-1 at 48 h as required, with cumulative values shown. Arrows indicate time point selected for microscopy analysis. Inset: qPCR of ATG5 or ATG7 cell lines 72 h after tetracy-cline induction (hatch) or control in BSF 2T1 clones expressing either YFP-ATG8.1 (grey) or YFP-ATG8.2 (black). Error bars represent one standard deviation derived from three replicates. (B) RNAi depletion of ATG genes influences autophagy. Constitutive expression of YFP-ATG8.1 and YFP-ATG8.2 in BSF 2T1 cells was visualised by fluorescent microscopy following individual RNAi of ATG5 and ATG7. Left hand images FITC filter set, right hand images FITC DIC merge. Scale bar 5 µm. (C) The mean number of autophagosomes per cell was determined in ATG5 and ATG7 RNAi lines by counting >200 cells 72 h after induction, with data displayed as a mean of three replicate experiments. Error bars represent standard deviation and asterisks indicate where data differed significantly from the mean of the un-induced controls **p<0.01. (D) Growth analysis of BSF ATG5 and CRK3 RNAi mutants in vivo. 1 x 105 (ATG5) and 5 x 105 (CRK3) trypanosomes were inoculated in 3 mice and RNAi induced with doxycycline (Dox) in 2 mice 24 h (ATG5) or 48 h (CRK3) later. Parasitaemia was monitored by tail bleed and counting on a haemocytometer. |
Having established that autophagy disruption does not impact BSF growth, we sought to investigate the response of ATG5 autophagy mutants to dihydroxyacetone (DHA). DHA is a trypanocidal compound that has been suggested to function by stimulating autophagic cell death; evidence for this is electron micrograph detection of vesicles reminiscent of autophagosomes post DHA treatment [29]. 1 mM DHA did not affect the number of observed YFP-ATG8.2 labelled autophagosomes, whereas 6 mM produced a mild yet significant increase in the number of autophagosomes per cell (Fig. 4A). To see whether the autophagy machinery was actively required for DHA-induced cell death, the IC50 for DHA against cells with and without a functional autophagy pathway were determined by inducing ATG5 RNAi 48 h pre-DHA incubation. This showed that there is no significant difference between the IC50 values for the un-induced control and ATG5 autophagy mutants (Fig. 4B), indicating that the autophagy machinery is not required for DHA-induced cell death in trypanosomes.
 | FIGURE 4: Autophagy is not required for dihydroxyacetone (DHA) induced bloodstream form cell death. (A) DHA increases autophagosome numbers in BSF 2T1 cells. BSF 2T1 cells expressing YFP-ATG8.2 were grown with 1 mM and 6 mM DHA for 20 h. The mean number of autophagosomes per cell was determined by counting >200 cells with data displayed as a mean of three replicate experiments and error bars represent standard deviation and asterisks indicate where data differed significantly from the mean of the untreated controls **p<0.001. (B) The IC50 of DHA was determined for BSF 2T1 YFP-ATG8.2 following 48 h ATG5 RNAi induction (tet+) and un-induced control cells (tet-). Parasites were cultured with serially dilut-ed DHA for 48 h before determining cell density. Graphs are representative of three replicate experiments. |
In Leishmania species autophagy facilitates lifecycle differentiation and thereby contributes to parasite virulence [12],[30]. Monomorphic BSF 2T1 parasites are not able to progress through the entire T. brucei lifecycle, however, stimulation with citrate/cis-aconitate and reduced incubation temperature to 27oC initiates differentiation to viable PCF parasites [31]. Accordingly, autophagy was disrupted with 48 h of ATG5 RNAi induction before stimulation of differentiation and analysis of characteristic PCF markers. The dynamics of EP-procyclin expression and repositioning of the kinetoplast in respect to the nucleus was unaffected by ATG5 RNAi (Fig. 5) indicating that autophagy is not required for BSF to PCF differentiation.
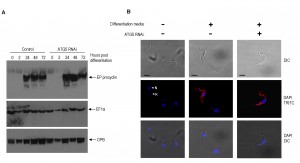 | FIGURE 5: Differentiation in bloodstream form RNAi autophagy mutants. Differentiation of BSF YFP-ATG8.2 ATG5 RNAi mutants (48 h after tet induction) was stimulated by switching cells to differentiation media and incubating at 27oC. Cells were analysed for characteristic PCF markers such as EP-procyclin expression and nuclear/kinetoplast re-configuration. (A) Western blot of whole cell extracts taken at specific times after induction of differentiation and probed with EP-procyclin, anti-EF1α and anti-OPB. (B) Fluorescent microscopy of cells 24 h post induction, fixed and labelled with anti-EP procyclin (red) and DAPI (blue) to show changes in the nuclear configuration (nucleus, N and kinetoplast, K). Scale bar 5 µm. |
–
Regulation of autophagy in BSF T. brucei by TOR
Although T. brucei contains an expanded TOR family (TOR1-4 [32]), rapamycin treatment of both BSF and PCF failed to significantly increase the number of YFP-ATG8.1 or YFP-ATG8.2 puncta (Fig. S4). This is in good agreement with findings for the PCF [16] and supports the atypical resistance displayed by the T. brucei TORC1 complex to rapamycin [22]. Nevertheless electron micrographs indicate that selective disruption of TORC1 through TOR1 RNAi can stimulate autophagy in BSF parasites [22]. To investigate this observation, the 2T1 autophagy reporter lines were used to analyse YFP-ATG8.1 and YFP-ATG8.2 labelled autophagosome formation following independent RNAi depletion of the four T. brucei TOR genes (Figs 6A and 6B). RNAi of the TOR orthologues produced differing effects on cell growth over the 96 h time course; depletion of TOR2 caused rapid cell death, while TOR1 and TOR4 depletion lead to substantial albeit less severe growth defects, and TOR3 depletion did not affect cell growth (Fig. 6A). Given such diversity, the time point selected to assess autophagosome formation was determined individually for each RNAi target, balancing the maximum time for target protein depletion with cell integrity. Autophagosome analysis was therefore carried out ahead of detected growth defects or at 72 h if no growth defect was observed (arrow heads Fig. 6A). To ensure valid assessment and comparison, the down-regulation of RNAi target transcript and YFP-ATG8 fusion protein expression post induction were confirmed for each autophagy analysis time point (Figs 6A and S5).
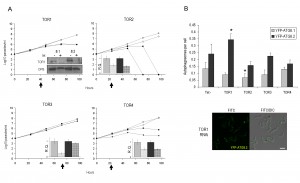 | FIGURE 6: RNAi of TOR genes in bloodstream form. (A) Growth analysis of BSF RNAi mutants in vitro. RNAi of TOR genes in BSF 2T1 constitutively expressing YFP-ATG8.1 (diamonds) and YFP-ATG8.2 (squares). RNAi induced with tetracycline (filled symbols) and growth compared to controls (empty symbols) for 96 h. Cells were reseeded to 1 x 104 ml-1 at 48 h as required, with cumulative growth shown. Arrows indicate time point selected for microscopy analysis following. Inset: confirmation of RNAi specificity by western blot (where antibody available, TOR1 [22]) or by qPCR of TOR RNAi cell lines at selected analysis time points after tetracycline induction (hatch) or control in BSF 2T1 clones expressing either YFP-ATG8.1 (grey) or YFP-ATG8.2 (black). Error bars represent one standard deviation derived from three replicates. (B) The mean number of autophagosomes per cell was determined in TOR RNAi lines by counting >200 cells at selected times after induction (arrows in A), with data displayed as a mean of at least three replicate experiments. Error bars represent standard deviation and asterisks indicate where data differed significantly from the mean of the un-induced controls *p<0.05. Constitutive expression of YFP-ATG8.2 in BSF 2T1 cells was visualised by fluorescent microscopy following individual RNAi of TOR1. Left hand images FITC filter set, right hand images FITC DIC merge. Scale bar 5 µm. |
In this study the only T. brucei TOR orthologue for which RNAi downregulation significantly increased the number of autophagosomes detected was TOR1, with RNAi increasing the YFP-ATG8.2 labelled autophagosomes per cell from 0.24 (±0.07) to 0.35 (±0.04) (Fig. 6B). However, only YFP-ATG8.2 was sensitive to TOR1 RNAi, as no significant change to YFP-ATG8.1 autophagosome formation was observed, pointing towards the presence of independent mechanisms of regulation for the two ATG8 orthologues. Furthermore, depletion of TOR2, TOR3 and TOR4 did not induce autophagosome formation; in fact, the only other significant change in comparison to the basal autophagosome levels was a slight decrease in YFP-ATG8.2 labelled autophagosomes following TOR2 RNAi (Fig. 6A). However, given the strong growth phenotype associated with TOR2 RNAi further detailed investigation is required to reveal the biological significance of this finding.
DISCUSSION
The widely established role of autophagy in the nutrient starvation response of many eukaryotes led us to initially investigate PCF T. brucei, which unlike the BSF are able to withstand periods of nutrient starvation. Our findings support the assertion that autophagy participates in a nutrient starvation response [16], with both YFP-ATG8.1 and YFP-ATG8.2 showing dramatic reorganisation into autophagosomes post starvation (Fig. 1). ATG8.3, however, despite significant similarity to ATG8.1 and ATG8.2 proteins, does not appear to play a functionally analogous role as during both normal growth and nutrient starvation YFP-ATG8.3 remains almost entirely dispersed in the cytosol. It is possible that ATG8.3 functions as a bone fide ATG8-like protein, but only in processes outside of the PCF nutrient starvation response. Such functional selectivity has been reported for ATG8.1 and ATG8.2 [16] and also occurs for the expanded L. major ATG8 family, with specific proteins linked to distinct autophagy processes [33]. However, an alternative hypothesis is that ATG8.3 functions as an ATG12. In support of this hypothesis, ATG8.3 is the syntenic orthologue of LmjF22.1300, a protein that despite some ATG8-like features, functions as an L. major ATG12 [30]. Furthermore, in yeast, the Atg12-Atg5 complex is predominantly located in the cytoplasm and associates only to the isolation membranes and not with mature autophagosomes [4]. Accordingly, the limited YFP-ATG8.3 puncta formation that occurs in response to PCF nutrient starvation in comparison to YFP-ATG8.1 or YFP–ATG8.2 could reflect a similar phenomenon. In the absence of definitive in vivo and in vitro biochemical data caution is still required with regard to definitively ascribing a function to ATG8.3 in T. brucei.
–
Although the application of molecular level analyses has begun to shed light on the mechanisms of T. brucei autophagy, its physiological role(s) remain largely unknown [16],[18]. To help address this issue we have developed and now validated BSF T. brucei autophagy reporter cell lines, in an RNAi compatible genetic background, that can be used to interrogate genes predicted to be involved in the autophagy pathway. The effectiveness of this system was demonstrated through the in vivo study of the T. brucei ATG5, ATG7 and the TOR family. Depletion of ATG5 following induction of ATG5-specific RNAi led to disruption of autophagosome formation, but this did not impact cell growth or the ability to differentiate under the conditions tested. The absence of detectable growth or differentiation phenotypes for the ATG5 RNAi mutant could, in part, be due to the highly virulent monomorphic BSF 2T1 line. The rapid growth rate of these cells might obscure the detection of subtle autophagy-linked defects under the experimental conditions tested. Future studies would benefit from creating ATG5 gene deletion mutants, or ATG5 RNAi in pleomorphic cell lines to enable the role of autophagy to be studied over the complete parasite life-cycle.
–
Analysis of the T. brucei TOR family successfully identified TOR1 as a negative regulator of autophagy in monomorphic BSF; controlling YFP-ATG8.2 puncta formation but not YFP-ATG8.1. However, further investigation is required to identify whether this discrimination relates to the potential hierarchical relationship of ATG8.1 and ATG8.2 in T. brucei autophagosome formation proposed to occur in the procyclic form [16] or reflects the existence of different signalling pathways that coordinate discreet subsets of autophagosomes. Interestingly, TOR4 has recently been shown to negatively regulate the differentiation of long slender BSF into the short stumpy stage pre-adapted for transmission to the insect host [32]. However, in this analysis RNAi of TOR4 did not increase the formation of autophagosomes (Fig. 6), although further analysis at additional time points might reveal a role for autophagy in this differentiation process.
–
The participation of autophagy in the PCF nutrient starvation response is consistent with the complex and prolonged differentiation processes that occur within the tsetse fly vector [34]. To ensure successful transmission to a mammalian host, the developing PCF parasites must be able to survive fluctuating nutrient stress attributed to inconsistent feeding patterns of tsetse flies in the wild [35]. It has been speculated that programmed cell death pathways, such as autophagic cell death, could function in altruistic mechanisms intended to limit the parasite burden placed upon the host, thereby promoting transmission of surviving parasites [16]. However, in the absence of conclusive evidence describing such pathways in T. brucei, it remains a distinct possibility that, as for many other eukaryotes, autophagy’s primary function is pro-survival [18]. Indeed, the reduced capacity of PCF autophagy mutants to recover from acute periods of nutrient starvation (Fig. 2), hints at how such a process could facilitate PCF survival during nutrient stress in the tsetse fly. Furthermore, the redundancy of the established BSF autophagy pathway during DHA-induced death (Fig. 4), highlights the difficulties in confidently attributing the autophagic machinery to an active cell death process and we believe that such an involvement has not been demonstrated to date for trypanosomatids [18].
MATERIALS AND METHODS
Parasite culture and genetic manipulation
Trypanosoma brucei Lister (strain 427) were grown in culture as bloodstream form (BSF) and procyclic form (PCF) parasites. BSF were maintained at 37°C in 5 % carbon dioxide, with wild type grown in HMI-9 media supplemented with 10 % heat-inactivated foetal calf serum (FCS), 10 % serum plus and 0.5 µg ml-1 penicillin-streptomycin (PS) (Sigma) and BSF 2T1 [25] grown in HMI-11 supplemented with tetracycline free FCS and 0.5 µg ml-1 PS. Wild type PCF and PCF 29-13 [24] T. brucei were grown at 27°C in 5 % carbon dioxide in SDM-79 media supplemented with 10 % FCS and 0.5 µg ml-1 PS (Sigma). Cell densities were determined by counting the number of cells in 10 μl of culture using an Improved Neubauer haemocytometer counting chamber (Weber Scientific).
–
For transfection, mid-log BSF (1 x 107) or PCF (3 x 107) cells were resuspended in 100 µl Human T Cell Nucleofector Solution (Lonza), transferred to a cuvette loaded with 10 µg of linearised DNA and pulsed on programme X-001 using the Human T Cell Nucleofector machine (Amaxa). The parasites were recovered overnight in HMI9, HMI11 or SDM79, respectively, before limited dilution cloning in appropriate media containing antibiotics for selection. BSF T. brucei 427: 5 μg ml-1 hygromycin B (Calbiochem), 2.5 μg ml-1 G418 (Calbiochem), 2.5 μg ml-1 phleomycin (InvivoGen) and 10 μg ml-1 blasticidin (Calbiochem). PCF T. brucei 427: 50 μg ml-1 hygromycin B (Calbiochem), 10 μg ml-1 G418 (Calbiochem), 10 μg ml-1 zeocin (Calbiochem) and 20 μg ml-1 blasticidin (Calbiochem).
–
RNAi was induced by adding 1 μg ml-1 tetracycline to cells at 1 x104 ml-1 (BSF) or 5 x 105 ml-1 (PCF) and the growth rate was compared to identical non-induced control lines. Cells were passaged every 24 h or 48 h (BSF) or 72 h (PCF) with tetracycline added to induced cells. For in vivo growth analysis 1 × 105 BSF parasites (ATG5) or 5 x 105 BSF parasites (CRK3) from a donor mouse were inoculated into 3 ICR mice by intraperitoneal injection. After 24 h (ATG5) or 48 h (CRK3), the drinking water of two mice was supplemented with 0.2g L-1 doxycycline with 5% sucrose. The parasitaemia in all mice was monitored daily by haemocytometer cell count of mouse blood taken from a tail vein diluted in 0.83% ammonium chloride. At parasitaemias above 1×108 cells per ml mice were humanely culled.
–
To monitor autophagy the number of YFP-ATG8 labelled autophagosomes per cell was determined by fluorescence microscopy of fixed cells. For each condition tested >200 cells were analysed, with data presented as a mean of at least three replicate experiments. Where required the conditional expression of YFP-ATG8 in PCF was induced by overnight incubation with 1 μg ml-1 tetracycline before further analysis. For nutrient starvation of PCF, the cells were washed twice and incubated in PBS warmed to 27oC, supplemented with wortmannin (Sigma) or 1 μg ml-1 tetracycline, as required.
–
For PCF starvation recovery assays, ATG3 and ATG7 RNAi were induced in cell lines expressing YFP-ATG8.1 for 72 h. The induced and control cells were washed twice in PBS and then either starved in PBS for 3.5 h or returned to SDM79, both supplemented with tetracycline as required. Cells were seeded at 1 x 106 ml-1 into fresh SDM79, with tetracycline as required, and growth was determined every 24 h by haemocytometer cell count for 96 h post starvation or mock SDM79 treatment.
–
For the phenotypic analysis of BSF autophagy mutants, ATG5 RNAi was induced in cells expressing YFP-ATG8.2 for 48 h to disrupt autophagy. To determine the IC50 of dihydroxyacetone (Sigma D107204) induced and control cells were counted, adjusted to 3 x 104 ml-1 in HMI-11 supplemented as required with 2 tetracycline μg ml-1 and 100 µl was added across a 96 well plate. Dihydroxyacteone was prepared to 24 mM in HMI-11 and 100 µl was serially diluted across the plate, with media only added to the last column. After 48 h cells were resuspended by gentle pipetting and growth determined by haemocytometer cell count of at least two replicate rows per treatment. The mean IC50 values were derived from three experimental replicates. To assess differentiation induced and control cells were counted, washed twice and adjusted to in 5 x 106 ml-1 in PCF differentiation media (SMD79, 10 mM glycerol, 6 mM cis-aconitate) and incubated at 27oC.
–
Plasmid Construction
The plasmids used for the ectopic expression of epitope tagged genes are described in detail by [36]. For the conditional expression of YFP-ATG8 fusion proteins ATG8.1 (Tb927.7.5900) was amplified from genomic DNA (strain 427) by PCR using primers OL2621 (HindIII) and OL2622 (BamHI); see table S1 for oligonucleotide sequences. The resulting fragment was cloned into p2628 [36], pre-digested with BamHI and HindIII. This created an N-terminal yellow fluorescent protein (YFP) tagged ATG8.1 expression construct, named pGL1843. Additional N-terminally tagged YFP expression constructs were prepared by following the same procedure for T. brucei ATG8.2 (Tb927.7.5910), using primers OL2623 (HindIII) and OL2624 (BamHI), producing pGL1835 and T. brucei ATG8.3 (Tb927.7.3320), using primers OL2625 (HindIII) and OL2626 (BamHI), producing pGL1836. Constructs were linearised with NotI for transfection. For constitutive ectopic expression of YFP-ATG8s, ORFs from pGL1843 and pGL1835 were cloned with HindIII and XhoI into a version of the T. brucei expression construct pGL2113 [26], that had been previously modified to change the drug resistance marker to BSD.
–
RNAi constructs. RNAi target fragments were identified using the TrypanoFAN: RNAit target selection script (http://trypanofan.path.cam.ac.uk/software/RNAit). For 2T7ti [27] constructs products were cloned with BamHI and HindIII using oligo pairs OL3668/OL3669 for ATG3 (Tb927.2.1890) creating pGL2065 and OL3670/OL3671 for ATG7 (Tb927.10.11180) creating pGL2066. To prepare the RNAi plasmids for transfection they were digested with NotI. For the stem-loop RNAi constructs, PCR amplified products were Gateway® cloned into pTL [28], an attP modified version of pRPISL [25]. This method was used to clone RNAi target fragments for: ATG5 (Tb927.6.2430) with oligos TL0413 and TL0414 creating pTL209; ATG7 (Tb927.10.11180) with oligos OL3810 and OL3811 creating pGL2171; and TOR1 (Tb927.10.8420) with oligos TL0297 and TL0298 creating pTL149, TOR2 (Tb927.4.420) with oligos TL0295 and TL0296 creating pTL148, TOR3 (Tb927.4.800) with oligos TL0294 and TL0295 creating pTL147 and TOR4 (Tb927.1.1930) with oligos TL0291 and TL0292 creating pTL146 [28]. The CRK3 (Tb927.10.4990) stem-loop RNAi construct pGL1987 was created by cloning the PCR product amplified with oligos OL3332 and OL3333 into pRPaiSL [25].
–
Antibodies
Antibodies were raised to recombinant T. brucei ATG8.1. ATG8.1 was amplified from genomic DNA (strain 427) using primers OL2775 (NheI) and OL2776 (XhoI). The product was cloned into the pET-28a(+) E. coli protein expression vector, pre-digested with NdeI and XhoI, generating pGL2115. pGL2155 was transformed into E. coli BL21 Rosetta (Stratagene) and ATG8.1 was expressed in 300 ml Overnight Express TB Medium (Novagen). The pellet was lysed with Bacterial Protein Extraction Reagent (B-PER) (Pierce) supplemented with 10 μg ml-1 DNAse 1 (Sigma). Recombinant HIS tagged protein was purified from the soluble fraction by immobilised metal ion affinity chromatography on a column packed with Metal Chelate-20 (Poros). Peak fractions were pooled and buffer exchanged into 50 mM Tris pH 8.0 then adjusted to pH 5.5 and 150 mM NaCl added. Purified recombinant ATG8.1 was used as an antigen to produce polyclonal antiserum in a rabbit (Scottish National Blood Transfusion Service, Penicuik, Midlothian). Antibodies were affinity purified using AminoLink Coupling Gel (Pierce) as per manufacturer’s guidelines.
–
Immunoblotting
Whole cell extracts were denatured by heating at 100oC for 4 min in LDS sample buffer (Invitrogen), and then electrophoresed on 12% or 15% (w/v) SDS-PAGE. Proteins were transferred to nitrocellulose membrane (Hybond-C, Amersham Biosciences) for western blotting. Primary antibodies rabbit anti-TbATG8.1, mouse anti-GFP (Living Colours), mouse anti-EF-1α (Millipore), rabbit anti-TbTOR1 [22] and sheep anti-oligopeptidase B [37] were used at 1:400, 1:800, 1:15000, 1:5000, respectively. HP-conjugated anti-mouse (Promega), anti-rabbit (Promega), and anti-sheep (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) were used at 1:5000. Chemiluminescent detection was with the SuperSignal West Pico substrate or West Femto Maximum Sensitivity Substrate (Pierce).
–
Microscopy
For each slide ~1 x 105 T. brucei cells were washed twice in trypanosome dilution buffer (TDB) (20 mM Na2HPO4, 2 mM NaH2PO4, 80 mM NaCl, 5 mM KCl, 1 mM MgSO4, 20 mM glucose, pH 7.4) before fixation in 1 % (w/v) paraformaldehyde (Sigma) on ice for 15 min (BSF) or 30 min (PCF). After two washes in TDB cells were resuspended in 20 µl TDB and evenly spread onto slides coated in 0.1 % poly-l-lysine (Sigma) and allowed to sediment. Where indicated DNA was visualised by staining with 1 μg ml-1 4,6-Diamindino-2-phenylindole (DAPI) in mounting solution (PBS, 50 % (v/v) glycerol, 2.5 % (w/v) DABCO). All slides were fitted with a coverslip sealed with nail varnish.
–
The number of autophagosomes per cell was determined using a Zeiss Axioplan microscope. Mean autophagosomes per cell were derived from >200 cells from at least 3 replicate experiments. Representative fluorescent microscopy images were obtained using an Applied Precision DeltaVision Deconvolution microscope system fitted with a CoolSnap HQ camera. Image capture and microscope operation was performed using the software package SoftWoRx. Fluorescence was viewed using the FITC filter (λEx 490 nm / λEm 528 nm), RD-TR-PE filter (λEx 555 nm / λEm 617 nm), DAPI filter (λEx 360 nm / λEm 457 nm) and reference images were obtained using the differential interference contrast (DIC) filter. Exposure time was set to 1 second with the transmission level of the neutral filter optimised for each sample and 60 x or 100 x oil immersion objectives were used. The Z-stacks obtained were deconvolved using the automatic conservative ratio setting before projection into one representative image. Merged images were created using Adobe Photoshop.
–
RNA isolation and quantitative PCR
RNA was isolated from 1.5 x 107 cells using the RNAeasy kit (Qiagen) processed by the QIAcube (Qiagen) according to manufacturer’s instructions, with an added on column DNase -1 (Sigma) digestion step. cDNA was produced from 1 μg of isolated RNA using random hexamer primers (Invitrogen) and SuperScript Reverse Transcriptase III (Invitrogen) according to manufacturers’ instructions. Genomic DNA contamination was controlled for by omitting reverse transcriptase (-RT). Quantitative real time PCR (qPCR) analysis was performed on cDNA samples in reactions comprised of following: 1 μl of cDNA, 12.5 μl Power SYBR Green PCR Master Mix (Applied Biosystems), 3 μM primer 1, 3 μM primer 2 and 6.5 μl of water. PCR reactions were carried out in triplicate in sealed 96 well plates, using an Applied Biosystems 7500 Real Time PCR machine, with resulting data analysed on Applied Biosystems 7500 System Software. Primers used for real time PCR were designed using the Applied Biosystems Primer Express 3.0 programme and designed to amplify ~50 bp product specific to each target ORF outside of the RNAi target region.
–
Statistical analysis of data
Where indicated data were expressed as means ± standard deviation from the mean. P-values were calculated using an unpaired, two-tailed Student’s T-test on Microsoft Excel. Differences were considered significant at a p-value of <0.05.
References
- D.J. Klionsky, F.C. Abdalla, H. Abeliovich, R.T. Abraham, A. Acevedo-Arozena, K. Adeli, L. Agholme, M. Agnello, P. Agostinis, J.A. Aguirre-Ghiso, H.J. Ahn, O. Ait-Mohamed, S. Ait-Si-Ali, T. Akematsu, S. Akira, H.M. Al-Younes, M.A. Al-Zeer, M.L. Albert, R.L. Albin, J. Alegre-Abarrategui, M.F. Aleo, M. Alirezaei, A. Almasan, M. Almonte-Becerril, A. Amano, R.K. Amaravadi, S. Amarnath, A.O. Amer, N. Andrieu-Abadie, V. Anantharam, D.K. Ann, S. Anoopkumar-Dukie, H. Aoki, N. Apostolova, G. Arancia, J.P. Aris, K. Asanuma, N.Y. Asare, H. Ashida, V. Askanas, D.S. Askew, P. Auberger, M. Baba, S.K. Backues, E.H. Baehrecke, B.A. Bahr, X. Bai, Y. Bailly, R. Baiocchi, G. Baldini, W. Balduini, A. Ballabio, B.A. Bamber, E.T. Bampton, G. Juhász, C.R. Bartholomew, D.C. Bassham, R.C. Bast, H. Batoko, B. Bay, I. Beau, D.M. Béchet, T.J. Begley, C. Behl, C. Behrends, S. Bekri, B. Bellaire, L.J. Bendall, L. Benetti, L. Berliocchi, H. Bernardi, F. Bernassola, S. Besteiro, I. Bhatia-Kissova, X. Bi, M. Biard-Piechaczyk, J.S. Blum, L.H. Boise, P. Bonaldo, D.L. Boone, B.C. Bornhauser, K.R. Bortoluci, I. Bossis, F. Bost, J. Bourquin, P. Boya, M. Boyer-Guittaut, P.V. Bozhkov, N.R. Brady, C. Brancolini, A. Brech, J.E. Brenman, A. Brennand, E.H. Bresnick, P. Brest, D. Bridges, M.L. Bristol, P.S. Brookes, E.J. Brown, J.H. Brumell, N. Brunetti-Pierri, U.T. Brunk, D.E. Bulman, S.J. Bultman, G. Bultynck, L.F. Burbulla, W. Bursch, J.P. Butchar, W. Buzgariu, S.P. Bydlowski, K. Cadwell, M. Cahová, D. Cai, J. Cai, Q. Cai, B. Calabretta, J. Calvo-Garrido, N. Camougrand, M. Campanella, J. Campos-Salinas, E. Candi, L. Cao, A.B. Caplan, S.R. Carding, S.M. Cardoso, J.S. Carew, C.R. Carlin, V. Carmignac, L.A. Carneiro, S. Carra, R.A. Caruso, G. Casari, C. Casas, R. Castino, E. Cebollero, F. Cecconi, J. Celli, H. Chaachouay, H. Chae, C. Chai, D.C. Chan, E.Y. Chan, R.C. Chang, C. Che, C. Chen, G. Chen, G. Chen, M. Chen, Q. Chen, S.S. Chen, W. Chen, X. Chen, X. Chen, X. Chen, Y. Chen, Y. Chen, Y. Chen, Y. Chen, Z. Chen, A. Cheng, C.H. Cheng, Y. Cheng, H. Cheong, J. Cheong, S. Cherry, R. Chess-Williams, Z.H. Cheung, E. Chevet, H. Chiang, R. Chiarelli, T. Chiba, L. Chin, S. Chiou, F.V. Chisari, C.H. Cho, D. Cho, A.M. Choi, D. Choi, K.S. Choi, M.E. Choi, S. Chouaib, D. Choubey, V. Choubey, C.T. Chu, T. Chuang, S. Chueh, T. Chun, Y. Chwae, M. Chye, R. Ciarcia, M.R. Ciriolo, M.J. Clague, R.S. Clark, P.G. Clarke, R. Clarke, P. Codogno, H.A. Coller, M.I. Colombo, S. Comincini, M. Condello, F. Condorelli, M.R. Cookson, G.H. Coombs, I. Coppens, R. Corbalan, P. Cossart, P. Costelli, S. Costes, A. Coto-Montes, E. Couve, F.P. Coxon, J.M. Cregg, J.L. Crespo, M.J. Cronjé, A.M. Cuervo, J.J. Cullen, M.J. Czaja, M. D'Amelio, A. Darfeuille-Michaud, L.M. Davids, F.E. Davies, M. De Felici, J.F. de Groot, C.A. de Haan, L. De Martino, A. De Milito, V. De Tata, J. Debnath, A. Degterev, B. Dehay, L.M. Delbridge, F. Demarchi, Y.Z. Deng, J. Dengjel, P. Dent, D. Denton, V. Deretic, S.D. Desai, R.J. Devenish, M. Di Gioacchino, G. Di Paolo, C. Di Pietro, G. Díaz-Araya, I. Díaz-Laviada, M.T. Diaz-Meco, J. Diaz-Nido, I. Dikic, S.P. Dinesh-Kumar, W. Ding, C.W. Distelhorst, A. Diwan, M. Djavaheri-Mergny, S. Dokudovskaya, Z. Dong, F.C. Dorsey, V. Dosenko, J.J. Dowling, S. Doxsey, M. Dreux, M.E. Drew, Q. Duan, M.A. Duchosal, K.E. Duff, I. Dugail, M. Durbeej, M. Duszenko, C.L. Edelstein, A.L. Edinger, G. Egea, L. Eichinger, N.T. Eissa, S. Ekmekcioglu, W.S. El-Deiry, Z. Elazar, M. Elgendy, L.M. Ellerby, K.E. Eng, A. Engelbrecht, S. Engelender, J. Erenpreisa, R. Escalante, A. Esclatine, E. Eskelinen, L. Espert, V. Espina, H. Fan, J. Fan, Q. Fan, Z. Fan, S. Fang, Y. Fang, M. Fanto, A. Fanzani, T. Farkas, J. Farre, M. Faure, M. Fechheimer, C.G. Feng, J. Feng, Q. Feng, Y. Feng, L. Fésüs, R. Feuer, M.E. Figueiredo-Pereira, G.M. Fimia, D.C. Fingar, S. Finkbeiner, T. Finkel, K.D. Finley, F. Fiorito, E.A. Fisher, P.B. Fisher, M. Flajolet, M.L. Florez-McClure, S. Florio, E.A. Fon, F. Fornai, F. Fortunato, R. Fotedar, D.H. Fowler, H.S. Fox, R. Franco, L.B. Frankel, M. Fransen, J.M. Fuentes, J. Fueyo, J. Fujii, K. Fujisaki, E. Fujita, M. Fukuda, R.H. Furukawa, M. Gaestel, P. Gailly, M. Gajewska, B. Galliot, V. Galy, S. Ganesh, B. Ganetzky, I.G. Ganley, F. Gao, G.F. Gao, J. Gao, L. Garcia, G. Garcia-Manero, M. Garcia-Marcos, M. Garmyn, A.L. Gartel, E. Gatti, M. Gautel, T.R. Gawriluk, M.E. Gegg, J. Geng, M. Germain, J.E. Gestwicki, D.A. Gewirtz, S. Ghavami, P. Ghosh, A.M. Giammarioli, A.N. Giatromanolaki, S.B. Gibson, R.W. Gilkerson, M.L. Ginger, H.N. Ginsberg, J. Golab, M.S. Goligorsky, P. Golstein, C. Gomez-Manzano, E. Goncu, C. Gongora, C.D. Gonzalez, R. Gonzalez, C. González-Estévez, R.A. González-Polo, E. Gonzalez-Rey, N.V. Gorbunov, S. Gorski, S. Goruppi, R.A. Gottlieb, D. Gozuacik, G.E. Granato, G.D. Grant, K.N. Green, A. Gregorc, F. Gros, C. Grose, T.W. Grunt, P. Gual, J. Guan, K. Guan, S.M. Guichard, A.S. Gukovskaya, I. Gukovsky, J. Gunst, �.B. Gustafsson, A.J. Halayko, A.N. Hale, S.K. Halonen, M. Hamasaki, F. Han, T. Han, M.K. Hancock, M. Hansen, H. Harada, M. Harada, S.E. Hardt, J.W. Harper, A.L. Harris, J. Harris, S.D. Harris, M. Hashimoto, J.A. Haspel, S. Hayashi, L.A. Hazelhurst, C. He, Y. He, M. Hébert, K.A. Heidenreich, M.H. Helfrich, G.V. Helgason, E.P. Henske, B. Herman, P.K. Herman, C. Hetz, S. Hilfiker, J.A. Hill, L.J. Hocking, P. Hofman, T.G. Hofmann, J. Höhfeld, T.L. Holyoake, M. Hong, D.A. Hood, G.S. Hotamisligil, E.J. Houwerzijl, M. Høyer-Hansen, B. Hu, C.A. Hu, H. Hu, Y. Hua, C. Huang, J. Huang, S. Huang, W. Huang, T.B. Huber, W. Huh, T. Hung, T.R. Hupp, G.M. Hur, J.B. Hurley, S.N. Hussain, P.J. Hussey, J.J. Hwang, S. Hwang, A. Ichihara, S. Ilkhanizadeh, K. Inoki, T. Into, V. Iovane, J.L. Iovanna, N.Y. Ip, Y. Isaka, H. Ishida, C. Isidoro, K. Isobe, A. Iwasaki, M. Izquierdo, Y. Izumi, P.M. Jaakkola, M. Jäättelä, G.R. Jackson, W.T. Jackson, B. Janji, M. Jendrach, J. Jeon, E. Jeung, H. Jiang, H. Jiang, J.X. Jiang, M. Jiang, Q. Jiang, X. Jiang, X. Jiang, A. Jiménez, M. Jin, S.V. Jin, C.O. Joe, T. Johansen, D.E. Johnson, G.V. Johnson, N.L. Jones, B. Joseph, S.K. Joseph, A.M. Joubert, G. Juhász, L. Juillerat-Jeanneret, C.H. Jung, Y. Jung, K. Kaarniranta, A. Kaasik, T. Kabuta, M. Kadowaki, K. Kågedal, Y. Kamada, V.O. Kaminskyy, H.H. Kampinga, H. Kanamori, C. Kang, K.B. Kang, K.I. Kang, R. Kang, Y. Kang, T. Kanki, T. Kanneganti, H. Kanno, A.G. Kanthasamy, A. Kanthasamy, V. Karantza, G.P. Kaushal, S. Kaushik, Y. Kawazoe, P. Ke, J.H. Kehrl, A. Kelekar, C. Kerkhoff, D.H. Kessel, H. Khalil, J.A. Kiel, A.A. Kiger, A. Kihara, D.R. Kim, D. Kim, D. Kim, E. Kim, H. Kim, J. Kim, J.H. Kim, J.C. Kim, J.K. Kim, P.K. Kim, S.W. Kim, Y. Kim, Y. Kim, A. Kimchi, A.C. Kimmelman, J.S. King, T.J. Kinsella, V. Kirkin, L.A. Kirshenbaum, K. Kitamoto, K. Kitazato, L. Klein, W.T. Klimecki, J. Klucken, E. Knecht, B.C. Ko, J.C. Koch, H. Koga, J. Koh, Y.H. Koh, M. Koike, M. Komatsu, E. Kominami, H.J. Kong, W. Kong, V.I. Korolchuk, Y. Kotake, M.I. Koukourakis, J.B.K. Flores, A.L. Kovács, C. Kraft, D. Krainc, H. Krämer, C. Kretz-Remy, A.M. Krichevsky, G. Kroemer, R. Krüger, O. Krut, N.T. Ktistakis, C. Kuan, R. Kucharczyk, A. Kumar, R. Kumar, S. Kumar, M. Kundu, H. Kung, T. Kurz, H.J. Kwon, A.R. La Spada, F. Lafont, T. Lamark, J. Landry, J.D. Lane, P. Lapaquette, J.F. Laporte, L. László, S. Lavandero, J.N. Lavoie, R. Layfield, P.A. Lazo, W. Le, L. Le Cam, D.J. Ledbetter, A.J. Lee, B. Lee, G.M. Lee, J. Lee, J. lee, M. Lee, M. Lee, S.H. Lee, C. Leeuwenburgh, P. Legembre, R. Legouis, M. Lehmann, H. Lei, Q. Lei, D.A. Leib, J. Leiro, J.J. Lemasters, A. Lemoine, M.S. Lesniak, D. Lev, V.V. Levenson, B. Levine, E. Levy, F. Li, J. Li, L. Li, S. Li, W. Li, X. Li, Y. Li, Y. Li, C. Liang, Q. Liang, Y. Liao, P.P. Liberski, A. Lieberman, H.J. Lim, K. Lim, K. Lim, C. Lin, F. Lin, J. Lin, J.D. Lin, K. Lin, W. Lin, W. Lin, Y. Lin, R. Linden, P. Lingor, J. Lippincott-Schwartz, M.P. Lisanti, P.B. Liton, B. Liu, C. Liu, K. Liu, L. Liu, Q.A. Liu, W. Liu, Y. Liu, Y. Liu, R.A. Lockshin, C. Lok, S. Lonial, B. Loos, G. Lopez-Berestein, C. López-Otín, L. Lossi, M.T. Lotze, P. Low, B. Lu, B. Lu, B. Lu, Z. Lu, F. Luciano, N.W. Lukacs, A.H. Lund, M.A. Lynch-Day, Y. Ma, F. Macian, J.P. MacKeigan, K.F. Macleod, F. Madeo, L. Maiuri, M.C. Maiuri, D. Malagoli, M.C.V. Malicdan, W. Malorni, N. Man, E. Mandelkow, S. Manon, I. Manov, K. Mao, X. Mao, Z. Mao, P. Marambaud, D. Marazziti, Y.L. Marcel, K. Marchbank, P. Marchetti, S.J. Marciniak, M. Marcondes, M. Mardi, G. Marfe, G. Mariño, M. Markaki, M.R. Marten, S.J. Martin, C. Martinand-Mari, W. Martinet, M. Martinez-Vicente, M. Masini, P. Matarrese, S. Matsuo, R. Matteoni, A. Mayer, N.M. Mazure, D.J. McConkey, M.J. McConnell, C. McDermott, C. McDonald, G.M. McInerney, S.L. McKenna, B. McLaughlin, P.J. McLean, C.R. McMaster, G.A. McQuibban, A.J. Meijer, M.H. Meisler, A. Meléndez, T.J. Melia, G. Melino, M.A. Mena, J.A. Menendez, R.F.S. Menna-Barreto, M.B. Menon, F.M. Menzies, C.A. Mercer, A. Merighi, D.E. Merry, S. Meschini, C.G. Meyer, T.F. Meyer, C. Miao, J. Miao, P.A. Michels, C. Michiels, D. Mijaljica, A. Milojkovic, S. Minucci, C. Miracco, C.K. Miranti, I. Mitroulis, K. Miyazawa, N. Mizushima, B. Mograbi, S. Mohseni, X. Molero, B. Mollereau, F. Mollinedo, T. Momoi, I. Monastyrska, M.M. Monick, M.J. Monteiro, M.N. Moore, R. Mora, K. Moreau, P.I. Moreira, Y. Moriyasu, J. Moscat, S. Mostowy, J.C. Mottram, T. Motyl, C.E. Moussa, S. Müller, S. Muller, K. Münger, C. Münz, L.O. Murphy, M.E. Murphy, A. Musarò, I. Mysorekar, E. Nagata, K. Nagata, A. Nahimana, U. Nair, T. Nakagawa, K. Nakahira, H. Nakano, H. Nakatogawa, M. Nanjundan, N.I. Naqvi, D.P. Narendra, M. Narita, M. Navarro, S.T. Nawrocki, T.Y. Nazarko, A. Nemchenko, M.G. Netea, T.P. Neufeld, P.A. Ney, I.P. Nezis, H.P. Nguyen, D. Nie, I. Nishino, C. Nislow, R.A. Nixon, T. Noda, A.A. Noegel, A. Nogalska, S. Noguchi, L. Notterpek, I. Novak, T. Nozaki, N. Nukina, T. Nürnberger, B. Nyfeler, K. Obara, T.D. Oberley, S. Oddo, M. Ogawa, T. Ohashi, K. Okamoto, N.L. Oleinick, F.J. Oliver, L.J. Olsen, S. Olsson, O. Opota, T.F. Osborne, G.K. Ostrander, K. Otsu, J.J. Ou, M. Ouimet, M. Overholtzer, B. Ozpolat, P. Paganetti, U. Pagnini, N. Pallet, G.E. Palmer, C. Palumbo, T. Pan, T. Panaretakis, U.B. Pandey, Z. Papackova, I. Papassideri, I. Paris, J. Park, O.K. Park, J.B. Parys, K.R. Parzych, S. Patschan, C. Patterson, S. Pattingre, J.M. Pawelek, J. Peng, D.H. Perlmutter, I. Perrotta, G. Perry, S. Pervaiz, M. Peter, G.J. Peters, M. Petersen, G. Petrovski, J.M. Phang, M. Piacentini, P. Pierre, V. Pierrefite-Carle, G. Pierron, R. Pinkas-Kramarski, A. Piras, N. Piri, L.C. Platanias, S. Pöggeler, M. Poirot, A. Poletti, C. Poüs, M. Pozuelo-Rubio, M. Prætorius-Ibba, A. Prasad, M. Prescott, M. Priault, N. Produit-Zengaffinen, A. Progulske-Fox, T. Proikas-Cezanne, S. Przedborski, K. Przyklenk, R. Puertollano, J. Puyal, S. Qian, L. Qin, Z. Qin, S.E. Quaggin, N. Raben, H. Rabinowich, S.W. Rabkin, I. Rahman, A. Rami, G. Ramm, G. Randall, F. Randow, V.A. Rao, J.C. Rathmell, B. Ravikumar, S.K. Ray, B.H. Reed, J.C. Reed, F. Reggiori, A. Régnier-Vigouroux, A.S. Reichert, J.J. Reiners, Jr., R.J. Reiter, J. Ren, J.L. Revuelta, C.J. Rhodes, K. Ritis, E. Rizzo, J. Robbins, M. Roberge, H. Roca, M.C. Roccheri, S. Rocchi, H.P. Rodemann, S. Rodríguez de Córdoba, B. Rohrer, I.B. Roninson, K. Rosen, M.M. Rost-Roszkowska, M. Rouis, K.M. Rouschop, F. Rovetta, B.P. Rubin, D.C. Rubinsztein, K. Ruckdeschel, E.B. Rucker, A. Rudich, E. Rudolf, N. Ruiz-Opazo, R. Russo, T.E. Rusten, K.M. Ryan, S.W. Ryter, D.M. Sabatini, J. Sadoshima, T. Saha, T. Saitoh, H. Sakagami, Y. Sakai, G.H. Salekdeh, P. Salomoni, P.M. Salvaterra, G. Salvesen, R. Salvioli, A.M. Sanchez, J.A. Sánchez-Alcázar, R. Sánchez-Prieto, M. Sandri, U. Sankar, P. Sansanwal, L. Santambrogio, S. Saran, S. Sarkar, M. Sarwal, C. Sasakawa, A. Sasnauskiene, M. Sass, K. Sato, M. Sato, A.H. Schapira, M. Scharl, H.M. Schätzl, W. Scheper, S. Schiaffino, C. Schneider, M.E. Schneider, R. Schneider-Stock, P.V. Schoenlein, D.F. Schorderet, C. Schüller, G.K. Schwartz, L. Scorrano, L. Sealy, P.O. Seglen, J. Segura-Aguilar, I. Seiliez, O. Seleverstov, C. Sell, J.B. Seo, D. Separovic, V. Setaluri, T. Setoguchi, C. Settembre, J.J. Shacka, M. Shanmugam, I.M. Shapiro, E. Shaulian, R.J. Shaw, J.H. Shelhamer, H. Shen, W. Shen, Z. Sheng, Y. Shi, K. Shibuya, Y. Shidoji, J. Shieh, C. Shih, Y. Shimada, S. Shimizu, T. Shintani, O.S. Shirihai, G.C. Shore, A.A. Sibirny, S.B. Sidhu, B. Sikorska, E.C. Silva-Zacarin, A. Simmons, A.K. Simon, H. Simon, C. Simone, A. Simonsen, D.A. Sinclair, R. Singh, D. Sinha, F.A. Sinicrope, A. Sirko, P.M. Siu, E. Sivridis, V. Skop, V.P. Skulachev, R.S. Slack, S.S. Smaili, D.R. Smith, M.S. Soengas, T. Soldati, X. Song, A.K. Sood, T.W. Soong, F. Sotgia, S.A. Spector, C.D. Spies, W. Springer, S.M. Srinivasula, L. Stefanis, J.S. Steffan, R. Stendel, H. Stenmark, A. Stephanou, S.T. Stern, C. Sternberg, B. Stork, P. Strålfors, C.S. Subauste, X. Sui, D. Sulzer, J. Sun, S. Sun, Z. Sun, J.J. Sung, K. Suzuki, T. Suzuki, M.S. Swanson, C. Swanton, S.T. Sweeney, L. Sy, G. Szabadkai, I. Tabas, H. Taegtmeyer, M. Tafani, K. Takács-Vellai, Y. Takano, K. Takegawa, G. Takemura, F. Takeshita, N.J. Talbot, K.S. Tan, K. Tanaka, K. Tanaka, D. Tang, D. Tang, I. Tanida, B.A. Tannous, N. Tavernarakis, G.S. Taylor, G.A. Taylor, J.P. Taylor, L.S. Terada, A. Terman, G. Tettamanti, K. Thevissen, C.B. Thompson, A. Thorburn, M. Thumm, F. Tian, Y. Tian, G. Tocchini-Valentini, A.M. Tolkovsky, Y. Tomino, L. Tönges, S.A. Tooze, C. Tournier, J. Tower, R. Towns, V. Trajkovic, L.H. Travassos, T. Tsai, M.P. Tschan, T. Tsubata, A. Tsung, B. Turk, L.S. Turner, S.C. Tyagi, Y. Uchiyama, T. Ueno, M. Umekawa, R. Umemiya-Shirafuji, V.K. Unni, M.I. Vaccaro, E.M. Valente, G. Van den Berghe, I.J. van der Klei, W.G. van Doorn, L.F. van Dyk, M. van Egmond, L.A. van Grunsven, P. Vandenabeele, W.P. Vandenberghe, I. Vanhorebeek, E.C. Vaquero, G. Velasco, T. Vellai, J.M. Vicencio, R.D. Vierstra, M. Vila, C. Vindis, G. Viola, M.T. Viscomi, O.V. Voitsekhovskaja, C. von Haefen, M. Votruba, K. Wada, R. Wade-Martins, C.L. Walker, C.M. Walsh, J. Walter, X. Wan, A. Wang, C. Wang, D. Wang, F. Wang, F. Wang, G. Wang, H. Wang, H. Wang, H. Wang, J. Wang, K. Wang, M. Wang, R.C. Wang, X. Wang, X.J. Wang, Y. Wang, Y. Wang, Z. Wang, Z.C. Wang, Z. Wang, D.G. Wansink, D.M. Ward, H. Watada, S.L. Waters, P. Webster, L. Wei, C.C. Weihl, W.A. Weiss, S.M. Welford, L. Wen, C.A. Whitehouse, J.L. Whitton, A.J. Whitworth, T. Wileman, J.W. Wiley, S. Wilkinson, D. Willbold, R.L. Williams, P.R. Williamson, B.G. Wouters, C. Wu, D. Wu, W.K. Wu, A. Wyttenbach, R.J. Xavier, Z. Xi, P. Xia, G. Xiao, Z. Xie, Z. Xie, D. Xu, J. Xu, L. Xu, X. Xu, A. Yamamoto, A. Yamamoto, S. Yamashina, M. Yamashita, X. Yan, M. Yanagida, D. Yang, E. Yang, J. Yang, S.Y. Yang, W. Yang, W.Y. Yang, Z. Yang, M. Yao, T. Yao, B. Yeganeh, W. Yen, J. Yin, X. Yin, O. Yoo, G. Yoon, S. Yoon, T. Yorimitsu, Y. Yoshikawa, T. Yoshimori, K. Yoshimoto, H.J. You, R.J. Youle, A. Younes, L. Yu, L. Yu, S. Yu, W.H. Yu, Z. Yuan, Z. Yue, C. Yun, M. Yuzaki, O. Zabirnyk, E. Silva-Zacarin, D. Zacks, E. Zacksenhaus, N. Zaffaroni, Z. Zakeri, H.J. Zeh, III, S.O. Zeitlin, H. Zhang, H. Zhang, J. Zhang, J. Zhang, L. Zhang, L. Zhang, M. Zhang, X.D. Zhang, M. Zhao, Y. Zhao, Y. Zhao, Z.J. Zhao, X. Zheng, B. Zhivotovsky, Q. Zhong, C. Zhou, C. Zhu, W. Zhu, X. Zhu, X. Zhu, Y. Zhu, T. Zoladek, W. Zong, A. Zorzano, J. Zschocke, and B. Zuckerbraun, "Guidelines for the use and interpretation of assays for monitoring autophagy", Autophagy, vol. 8, pp. 445-544, 2012. http://dx.doi.org/10.4161/auto.19496
- Z. Yang, and D.J. Klionsky, "An Overview of the Molecular Mechanism of Autophagy", Current Topics in Microbiology and Immunology, pp. 1-32, 2009. http://dx.doi.org/10.1007/978-3-642-00302-8_1
- C. He, and D.J. Klionsky, "Regulation Mechanisms and Signaling Pathways of Autophagy", Annual Review of Genetics, vol. 43, pp. 67-93, 2009. http://dx.doi.org/10.1146/annurev-genet-102808-114910
- N. Mizushima, A. Yamamoto, M. Hatano, Y. Kobayashi, Y. Kabeya, K. Suzuki, T. Tokuhisa, Y. Ohsumi, and T. Yoshimori, "Dissection of Autophagosome Formation Using Apg5-Deficient Mouse Embryonic Stem Cells", The Journal of Cell Biology, vol. 152, pp. 657-668, 2001. http://dx.doi.org/10.1083/jcb.152.4.657
- N. Mizushima, T. Noda, T. Yoshimori, Y. Tanaka, T. Ishii, M.D. George, D.J. Klionsky, M. Ohsumi, and Y. Ohsumi, "A protein conjugation system essential for autophagy", Nature, vol. 395, pp. 395-398, 1998. http://dx.doi.org/10.1038/26506
- Y. Ichimura, T. Kirisako, T. Takao, Y. Satomi, Y. Shimonishi, N. Ishihara, N. Mizushima, I. Tanida, E. Kominami, M. Ohsumi, T. Noda, and Y. Ohsumi, "A ubiquitin-like system mediates protein lipidation", Nature, vol. 408, pp. 488-492, 2000. http://dx.doi.org/10.1038/35044114
- K. Suzuki, Y. Kubota, T. Sekito, and Y. Ohsumi, "Hierarchy of Atg proteins in pre‐autophagosomal structure organization", Genes to Cells, vol. 12, pp. 209-218, 2007. http://dx.doi.org/10.1111/j.1365-2443.2007.01050.x
- N. Mizushima, T. Noda, and Y. Ohsumi, "Apg16p is required for the function of the Apg12p–Apg5p conjugate in the yeast autophagy pathway", The EMBO Journal, vol. 18, pp. 3888-3896, 1999. http://dx.doi.org/10.1093/emboj/18.14.3888
- J. Romanov, M. Walczak, I. Ibiricu, S. Schüchner, E. Ogris, C. Kraft, and S. Martens, "Mechanism and functions of membrane binding by the Atg5-Atg12/Atg16 complex during autophagosome formation", The EMBO Journal, vol. 31, pp. 4304-4317, 2012. http://dx.doi.org/10.1038/emboj.2012.278
- Y. Ohsumi, and N. Mizushima, "Two ubiquitin-like conjugation systems essential for autophagy", Seminars in Cell & Developmental Biology, vol. 15, pp. 231-236, 2004. http://dx.doi.org/10.1016/j.semcdb.2003.12.004
- D.J. Rigden, P.A. Michels, and M.L. Ginger, "Autophagy in protists: Examples of secondary loss, lineage-specific innovations, and the conundrum of remodeling a single mitochondrion.", Autophagy, 2009. http://www.ncbi.nlm.nih.gov/pubmed/19483474
- R.A. Williams, L. Tetley, J.C. Mottram, and G.H. Coombs, "Cysteine peptidases CPA and CPB are vital for autophagy and differentiation in Leishmania mexicana", Molecular Microbiology, vol. 61, pp. 655-674, 2006. http://dx.doi.org/10.1111/j.1365-2958.2006.05274.x
- M. Herman, S. Gillies, P.A. Michels, and D.J. Rigden, "Autophagy and related processes in trypanosomatids: insights from genomic and bioinformatic analyses.", Autophagy, 2006. http://www.ncbi.nlm.nih.gov/pubmed/16874069
- A.M. Tomlins, F. Ben-Rached, R.A. Williams, W.R. Proto, I. Coppens, U. Ruch, T.W. Gilberger, G.H. Coombs, J.C. Mottram, S. Müller, and G. Langsley, "Plasmodium falciparumATG8 implicated in both autophagy and apicoplast formation", Autophagy, vol. 9, pp. 1540-1552, 2013. http://dx.doi.org/10.4161/auto.25832
- M. Duszenko, M.L. Ginger, A. Brennand, M. Gualdrón-López, M.I. Colombo, G.H. Coombs, I. Coppens, B. Jayabalasingham, G. Langsley, S. Lisboa de Castro, R. Menna-Barreto, J.C. Mottram, M. Navarro, D.J. Rigden, P.S. Romano, V. Stoka, B. Turk, and P.A. Michels, "Autophagy in protists", Autophagy, vol. 7, pp. 127-158, 2011. http://dx.doi.org/10.4161/auto.7.2.13310
- F. Li, Q. Shen, C. Wang, Y. Sun, A.Y. Yuan, and C.Y. He, "A role of autophagy in Trypanosoma brucei cell death", Cellular Microbiology, vol. 14, pp. 1242-1256, 2012. http://dx.doi.org/10.1111/j.1462-5822.2012.01795.x
- D. Ghosh, J.L. Walton, P.D. Roepe, and A.P. Sinai, "Autophagy is a cell death mechanism in Toxoplasma gondii", Cellular Microbiology, vol. 14, pp. 589-607, 2012. http://dx.doi.org/10.1111/j.1462-5822.2011.01745.x
- W.R. Proto, G.H. Coombs, and J.C. Mottram, "Cell death in parasitic protozoa: regulated or incidental?", Nature Reviews Microbiology, vol. 11, pp. 58-66, 2012. http://dx.doi.org/10.1038/nrmicro2929
- S. Wullschleger, R. Loewith, and M.N. Hall, "TOR Signaling in Growth and Metabolism", Cell, vol. 124, pp. 471-484, 2006. http://dx.doi.org/10.1016/j.cell.2006.01.016
- X. Zheng, D. Fiorentino, J. Chen, G.R. Crabtree, and S.L. Schreiber, "TOR kinase domains are required for two distinct functions, only one of which is inhibited by rapamycin", Cell, vol. 82, pp. 121-130, 1995. http://dx.doi.org/10.1016/0092-8674(95)90058-6
- Y. Kamada, T. Funakoshi, T. Shintani, K. Nagano, M. Ohsumi, and Y. Ohsumi, "Tor-Mediated Induction of Autophagy via an Apg1 Protein Kinase Complex", The Journal of Cell Biology, vol. 150, pp. 1507-1513, 2000. http://dx.doi.org/10.1083/jcb.150.6.1507
- A. Barquilla, J.L. Crespo, and M. Navarro, "Rapamycin inhibits trypanosome cell growth by preventing TOR complex 2 formation", Proceedings of the National Academy of Sciences, vol. 105, pp. 14579-14584, 2008. http://dx.doi.org/10.1073/pnas.0802668105
- R. Koopmann, K. Muhammad, M. Perbandt, C. Betzel, and M. Duszenko, "Trypanosoma bruceiATG8: Structural insights into autophagic-like mechanisms in protozoa", Autophagy, vol. 5, pp. 1085-1091, 2009. http://dx.doi.org/10.4161/auto.5.8.9611
- E. Wirtz, S. Leal, C. Ochatt, and G. Cross, "A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei", Molecular and Biochemical Parasitology, vol. 99, pp. 89-101, 1999. http://dx.doi.org/10.1016/S0166-6851(99)00002-X
- S. Alsford, and D. Horn, "Single-locus targeting constructs for reliable regulated RNAi and transgene expression in Trypanosoma brucei", Molecular and Biochemical Parasitology, vol. 161, pp. 76-79, 2008. http://dx.doi.org/10.1016/j.molbiopara.2008.05.006
- E. Myburgh, J.A. Coles, R. Ritchie, P.G.E. Kennedy, A.P. McLatchie, J. Rodgers, M.C. Taylor, M.P. Barrett, J.M. Brewer, and J.C. Mottram, "In Vivo Imaging of Trypanosome-Brain Interactions and Development of a Rapid Screening Test for Drugs against CNS Stage Trypanosomiasis", PLoS Neglected Tropical Diseases, vol. 7, pp. e2384, 2013. http://dx.doi.org/10.1371/journal.pntd.0002384
- D.J. LaCount, S. Bruse, K.L. Hill, and J.E. Donelson, "Double-stranded RNA interference in Trypanosoma brucei using head-to-head promoters", Molecular and Biochemical Parasitology, vol. 111, pp. 67-76, 2000. http://dx.doi.org/10.1016/S0166-6851(00)00300-5
- N. Jones , E. Thomas, E. Brown, N. Dickens, T. Hammarton, and J. Mottram, "Regulators of Trypanosoma brucei cell cycle progression and differentiation identified using a kinome-wide RNAi screen", PLoS Path, in press, 2013.
- N.L. Uzcátegui, V. Denninger, P. Merkel, C. Schoenfeld, K. Figarella, and M. Duszenko, "Dihydroxyacetone induced autophagy in African trypanosomes.", Autophagy, 2007. http://www.ncbi.nlm.nih.gov/pubmed/17786028
- R.A.M. Williams, T.K. Smith, B. Cull, J.C. Mottram, and G.H. Coombs, "ATG5 Is Essential for ATG8-Dependent Autophagy and Mitochondrial Homeostasis in Leishmania major", PLoS Pathogens, vol. 8, pp. e1002695, 2012. http://dx.doi.org/10.1371/journal.ppat.1002695
- K. ZIEGELBAUER, M. QUINTEN, H. SCHWARZ, T.W. PEARSON, and P. OVERATH, "Synchronous differentiation of Trypanosoma brucei from bloodstream to procyclic forms in vitro", European Journal of Biochemistry, vol. 192, pp. 373-378, 1990. http://dx.doi.org/10.1111/j.1432-1033.1990.tb19237.x
- A. Barquilla, M. Saldivia, R. Diaz, J. Bart, I. Vidal, E. Calvo, M.N. Hall, and M. Navarro, "Third target of rapamycin complex negatively regulates development of quiescence in Trypanosoma brucei", Proceedings of the National Academy of Sciences, vol. 109, pp. 14399-14404, 2012. http://dx.doi.org/10.1073/pnas.1210465109
- R.A. Williams, K.L. Woods, L. Juliano, J.C. Mottram, and G.H. Coombs, "Characterization of unusual families of ATG8-like proteins and ATG12 in the protozoan parasiteLeishmania major", Autophagy, vol. 5, pp. 159-172, 2009. http://dx.doi.org/10.4161/auto.5.2.7328
- J. Van Den Abbeele, Y. Claes, D. van Bockstaele, D. Le Ray, and M. Coosemans, "Trypanosoma brucei spp. development in the tsetse fly: characterization of the post-mesocyclic stages in the foregut and proboscis.", Parasitology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10363280
- S. Aksoy, W.C. Gibson, and M.J. Lehane, "Interactions between tsetse and trypanosomes with implications for the control of trypanosomiasis", Advances in Parasitology, pp. 1-83, 2003. http://dx.doi.org/10.1016/S0065-308X(03)53002-0
- S. Kelly, J. Reed, S. Kramer, L. Ellis, H. Webb, J. Sunter, J. Salje, N. Marinsek, K. Gull, B. Wickstead, and M. Carrington, "Functional genomics in Trypanosoma brucei: A collection of vectors for the expression of tagged proteins from endogenous and ectopic gene loci", Molecular and Biochemical Parasitology, vol. 154, pp. 103-109, 2007. http://dx.doi.org/10.1016/j.molbiopara.2007.03.012
- J.C. Munday, K. McLuskey, E. Brown, G.H. Coombs, and J.C. Mottram, "Oligopeptidase B deficient mutants of Leishmania major", Molecular and Biochemical Parasitology, vol. 175, pp. 49-57, 2011. http://dx.doi.org/10.1016/j.molbiopara.2010.09.003
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
This work was supported by an MRC studentship to WRP and MRC programme grants (G0700127 and MR/K019384/1) to JCM and GHC. The Welcome Trust Centre for Molecular Parasitology is supported by core funding from the Welcome Trust (085349). We thank Elaine Brown and Alana Hamilton for assistance culturing parasites and Miguel Navarro for the anti-TOR1 antibody.
COPYRIGHT
© 2013

Tracking autophagy during proliferation and differentiation of Trypanosoma brucei by William R. Proto et al. is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.








