
Research Articles:
Microbial Cell, Vol. 5, No. 5, pp. 220 - 232; doi: 10.15698/mic2018.05.629
Decreasing cytosolic translation is beneficial to yeast and human Tafazzin-deficient cells

1 Institut de Biochimie et Génétique Cellulaires, CNRS UMR 5095, Université de Bordeaux, 1 rue Camille Saint-Saëns, 33077 Bordeaux cedex, France.
2 Laboratoire de Biogenèse Membranaire, CNRS UMR 5200, Université de Bordeaux, INRA Bordeaux Aquitaine, Villenave d’Ornon, France.
3 LEB Aquitaine Transfert-ADERA, FR-33883 Villenave d’Ornon, Cedex, France.
4 Institute of Biochemistry and Biophysics, Polish Academy of Sciences, Warsaw, Poland.
5 CNRS FR3636 Fédération de recherché en Neuroscience, Université Paris-Descartes, 45, rue des Saints-Pères, 75006 Paris, France.
# Research associate from INSERM.
$ These authors contributed equally.
Keywords: mitochondrial disease, oxidative phosphorylation, Barth syndrome, cytosolic protein synthesis, cycloheximide; cardiolipin remodeling.
Abbreviations:
BTHS – Barth syndrome,
CHX – cycloheximide,
CL – cardiolipin,
OXPHOS – oxidative phosphorylation,
ROS – reactive oxygen species,
WT –wild type.
Received originally: 03/11/2017 Received in revised form: 01/02/2018
Accepted: 05/02/2018
Published: 18/02/2018
Correspondence:
Déborah Tribouillard-Tanvier, Phone: +33 5 56 99 90 39, Fax: +33 5 56 99 90 51; deborah.tribouillard-tanvier@ibgc.cnrs.fr
Jean-Paul Lasserre, Phone: +33 5 56 99 90 39, Fax: +33 5 56 99 90 51; jean-paul.lasserre@ibgc.cnrs.fr
Conflict of interest statement: The authors declare no competing or financial interests.
Please cite this article as: Maxence de Taffin de Tilques, Jean-Paul Lasserre, François Godard, Elodie Sardin, Marine Bouhier, Marina Le Guedard, Roza Kucharczyk, Patrice X. Petit, Eric Testet, Jean-Paul di Rago, Déborah Tribouillard-Tanvier (2018). Decreasing cytosolic translation is beneficial to yeast and human Tafazzin-deficient cells. Microbial Cell 5(5): 220-232. doi: 10.15698/mic2018.05.629
Abstract
Cardiolipin (CL) optimizes diverse mitochondrial processes, including oxidative phosphorylation (OXPHOS). To function properly, CL needs to be unsaturated, which requires the acyltransferase Tafazzin (TAZ). Loss-of-function mutations in the TAZ gene are responsible for the Barth syndrome (BTHS), a rare X-linked cardiomyopathy, presumably because of a diminished OXPHOS capacity. Herein we show that a partial inhibition of cytosolic protein synthesis, either chemically with the use of cycloheximide or by specific genetic mutations, fully restores biogenesis and the activity of the oxidative phosphorylation system in a yeast BTHS model (taz1Δ). Interestingly, the defaults in CL were not suppressed, indicating that they are not primarily responsible for the OXPHOS deficiency in taz1Δ yeast. Low concentrations of cycloheximide in the picomolar range were beneficial to TAZ-deficient HeLa cells, as evidenced by the recovery of a good proliferative capacity. These findings reveal that a diminished capacity of CL remodeling deficient cells to preserve protein homeostasis is likely an important factor contributing to the pathogenesis of BTHS. This in turn, identifies cytosolic translation as a potential therapeutic target for the treatment of this disease.
INTRODUCTION
The Barth Syndrome (BTHS) is a rare X-linked recessive mitochondrial disorder that is characterized by cardiac and skeletal myopathies, growth retardation, hypocholesterolemia, increased urine levels of 3-methylglutaconic acid and high susceptibility to bacterial infections, due to cyclic neutropenia [1][2][3]. This disease is caused by mutations in Tafazzin, a mitochondrial protein involved in the remodeling of cardiolipin (CL). This phospholipid is mainly found in mitochondria, [4][5][6][7][8][9][10] where it optimizes numerous processes including oxidative phosphorylation (OXPHOS) [11][12][13], fusion [14], fission [15][16], protein import [17][18], iron-sulfur cluster biogenesis [19], mitophagy [20][21][22][23], apoptosis [7][23][24][25][26][27][28] and the transport of metabolites across the mitochondrial inner membrane [6][17][29][30][31][32][33][34][35][36]. Tafazzin is an acyltransferase required for the maintenance of unsaturated carbon-carbon bonds in CL fatty acyl chains [1][37][38][39][40][41]. Loss-of-function mutations in Tafazzin lead to reduced levels of unsaturated CL and the accumulation of CL species with an incomplete set of fatty acyl chains (such as monolysocardiolipin, MLCL) [42][43][44]. This in turn results in multiple mitochondrial alterations that ultimately compromise the OXPHOS capacity [24][45][46][47][48].
–
Simple model organisms such as Saccharomyces cerevisiae or baker’s yeast are an important resource for the study of mitochondrial diseases. Mitochondria from this single-celled fungus and humans show many similarities [49][50][51][52][53]. Being easily amenable to genetic manipulation of mitochondrial function [54][55], and owing to the ability of yeast to survive the loss of oxidative phosphorylation; yeast models of human mitochondrial diseases can be easily created and kept alive when provided with fermentable substrates [56][57]. The common respiratory growth defect of these models enables large-scale screening of genetic and pharmacological suppressors [57][58][59]. Yeast has in this way already pointed to several potential druggable therapeutic intervention points, such as the oxodicarboxylic acid carrier [60] and mitochondrial protein import [61], among others.
–
Herein we report that reducing cytosolic protein synthesis preserves OXPHOS in CL remodeling deficient yeast and improves the growth rate and viability of human HeLa cells lacking Tafazzin. This study sheds new light on the pathogenesis of BTHS and identifies cytosolic protein synthesis as a potential intervention point for the treatment of the disease.
RESULTS
Decreasing cytosolic protein synthesis improves respiratory growth of taz1Δ yeast
We [60] and others [62] showed that yeast cells lacking the gene encoding Taz1p (taz1Δ) grow poorly on respiratory carbon sources at 36°C, compared to the wild-type (WT) TAZ1+ strain. Using a drug screening procedure we previously described [58], we found that cytosolic protein synthesis inhibitors such as, cycloheximide (hereafter abbreviated as CHX), anisomycin and emetine suppressed this phenotype in a dose-dependent manner. In the tests shown in Fig. 1A, taz1Δ cells freshly grown by fermentation in glucose were spread on plates containing ethanol (respiratory carbon source) and then exposed to paper disks spotted with the drugs dissolved in DMSO. After five days of incubation at 36°C, halos of enhanced growth appeared around the filters, whereas DMSO alone had no effect. In this assay the compounds diffused into the medium, explaining why growth was improved only at some distance around the filters, below which it was totally absent due to a too high concentration of the protein synthesis inhibitors. CHX was active at 20-30 fold lower concentrations compared to anisomycin and emetine (Fig. 1A). The optimal rescuing concentration range of CHX was determined by growth tests in liquid media containing 2% ethanol and 0.5% galactose, at 36°C. After consumption of the galactose, which is a fermentable substrate, growth of taz1Δ yeast was much less efficient compared to the WT, owing to its failure to properly express mitochondrial function (Fig. 1B). The best growth improvement of taz1Δ yeast was observed in the presence of 10 nM CHX. At this concentration, growth of the wild type was unaffected (Fig. 1B). Pulse labeling of proteins with S35-methionine and S35-cysteine revealed that the rate of cytosolic protein synthesis was decreased by about 50% in taz1Δ yeast grown in the presence of 10 nM CHX, in comparison to the WT (Fig. 1C). Interestingly, cytosolic translation was already decreased in the mutant grown in the absence of the drug by about 35%, possibly as a means to attenuate a protein stress induced by a lack in CL remodeling (see below).
–
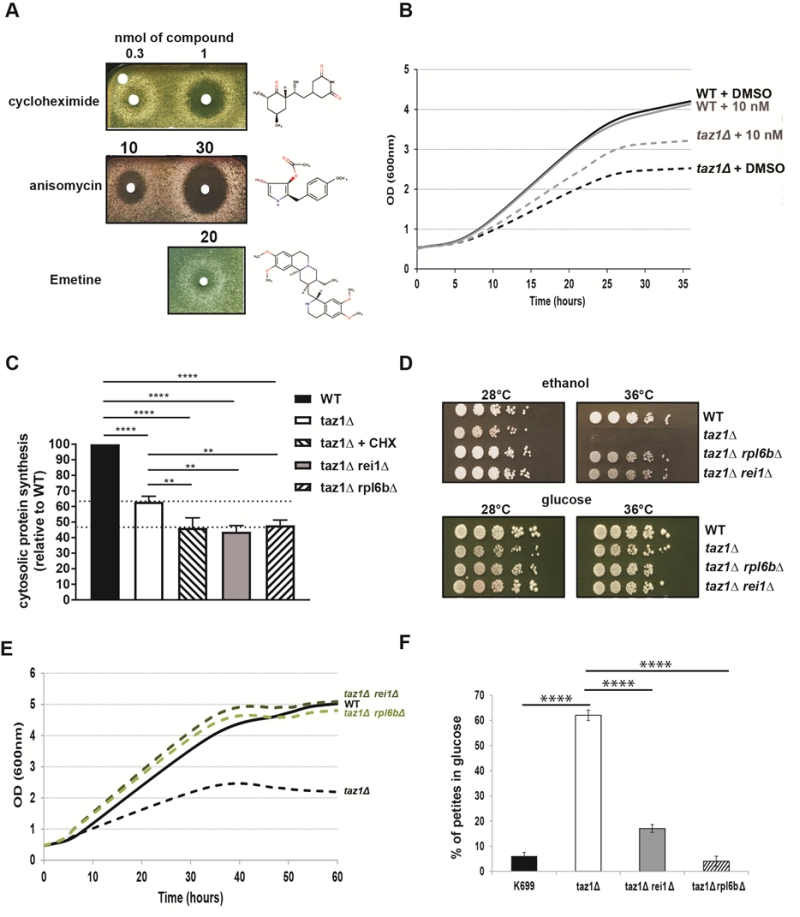 |
FIGURE 1: Partially decreasing cytosolic translation in Taffazin-deficient (taz1Δ) yeast improves respiration-dependent growth and mtDNA maintenance. (A) taz1Δ yeast cells were spread as dense layers onto rich ethanol solid media and then exposed to sterile filters spotted with cycloheximide, anisomycin or emetine (dissolved in DMSO). The plates were scanned after 5 days of incubation at 36°C. The filter at the top left was spotted with DMSO alone to provide a negative control. (B) Determination in liquid cultures of CHX concentrations that optimally rescue taz1Δ yeast. Complete synthetic media (CSM) containing 0.5% galactose + 2% ethanol sup-plemented or not with CHX at the indicated concentrations were inoculated with WT and taz1Δ cells pre-grown in CSM containing 2% glucose at 28°C. The cultures were performed at 36°C and cells densities (OD600nm) taken over a period of 36 hours. (C) Rate of cytosolic protein synthesis. Total proteins and mitochondrial proteins were labeled with a mixture of [35S]-methionine and [35S]-cysteine for 20 min in whole cells from wild type, taz1Δ rei1Δ, taz1Δ rpl6bΔ and taz1Δ yeast grown for 24 hours in rich 0.5% galactose + 2% ethanol at 36°C, and taz1Δ cells grown in the same conditions in presence of 10 nM cycloheximide (CHX). After the labeling reactions, total protein extracts were prepared and separated by SDS-PAGE on a 12% polyacrylamide gel (75 µg per lane). The gels were dried and analyzed with a PhosphorImager. Quantification was performed using Image J. Data are expressed in % relative to the WT (n=3). The shown data are cytosolic protein synthesis rates (total minus mitochondrial protein synthesis rates). Statistical analysis was done with Tukey’s test (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001). (D) Genetic ablation of REI1 (rei1Δ) or RPL6B (rpl6bΔ) improves respiratory growth of taz1Δ yeast. WT, taz1Δ, taz1Δ rei1Δ and taz1Δ rpl6bΔ cells freshly grown at 28°C in rich glucose were serially diluted and spotted onto rich ethanol and glucose plates. The plates were scanned after 4 days of incubation at the indicated temperature. (E) Growth of WT, taz1Δ, taz1Δ rei1Δ and taz1Δ rpl6bΔ strains in liquid complete synthetic media containing 0.5% galactose + 2% ethanol at 36°C. The cultures were inoculated with cells grown in CSM containing 2% glucose at 28°C. The cultures were performed at 36°C and cell densities (OD600nm) taken over a period of 60 hours. (F) Genetic ablation of REI1 (rei1Δ) or RPL6B (rpl6bΔ) in taz1Δ yeast preserves mtDNA maintenance. Proportions of ρ-/ρ0 cells produced in glucose cultures at 28°C of strains WT, taz1Δ, taz1Δ reiΔ, and taz1Δ rpl6bΔ were determined using the procedure described in [60] (n=3). Data are expressed in % relative to the WT and were statistically analyzed using Tukey’s test (*P<0.05; **P<0.01; ***P<0.001). |
If CHX is a well-known inhibitor of cytosolic translation, one cannot exclude that it has other effects in cells that could be responsible for the improved respiratory growth of taz1Δ yeast. We therefore tested the effects on taz1Δ yeast of null mutations in the genes REI1 and RPL6B that are known to partially inhibit cytosolic protein synthesis by 20% and 30% respectively [63][64]. The double mutants taz1Δ rei1Δ and taz1Δ rpl6bΔ grew efficiently on respiratory carbon sources (Fig. 1D, E), and showed a 50% drop in the rate of protein synthesis (Fig. 1C). These data confirmed that the beneficial effect of CHX in taz1Δ yeast resulted from a decreased rate of protein synthesis.
–
Decreasing cytosolic protein synthesis improves mtDNA maintenance in taz1Δyeast
We previously showed that taz1Δ yeast grown by fermentation at 28°C, i.e. in conditions where the presence of functional mtDNA is not indispensable, has an increased propensity to produce ρ–/ρ0 cells issued from large deletions in the mitochondrial genome (60% vs 5% in the WT) [60]. The double mutants taz1Δ rei1Δ and taz1Δ rpl6bΔ produced five to ten times less ρ–/ρ0 cells than taz1Δ yeast in glucose cultures (Fig. 1F). Thus, partially decreasing cytosolic translation preserves a proper maintenance of mtDNA in CL deficient yeast cells.
–
Reducing cytosolic translation does not restore CL remodeling in taz1Δ yeast
As reported [39][60][62], mitochondria from taz1Δ yeast, compared to those from the WT, have 50% less CL, a 2-fold higher content in phosphatidylinositol (PI), whereas phosphatidylethanolamine (PE) and phosphatidylcholine (PC) accumulated normally (Fig. 2A). Additionally, the remaining CL species are less unsaturated as suggested by the decreased levels in oleic acid chains (C18:1) and increased stearic (C18:0) and palmitic (C16:0) groups compared to CL molecules extracted from the WT (Fig. 2B). Strains taz1Δ rei1Δ and taz1Δ rpl6bΔ showed very similar phospholipid profiles (Fig. 2A, B), indicating that mitochondrial function recovery in taz1Δ yeast upon partial inhibition of cytosolic translation did not result from an enhanced production of mature CL species.
–
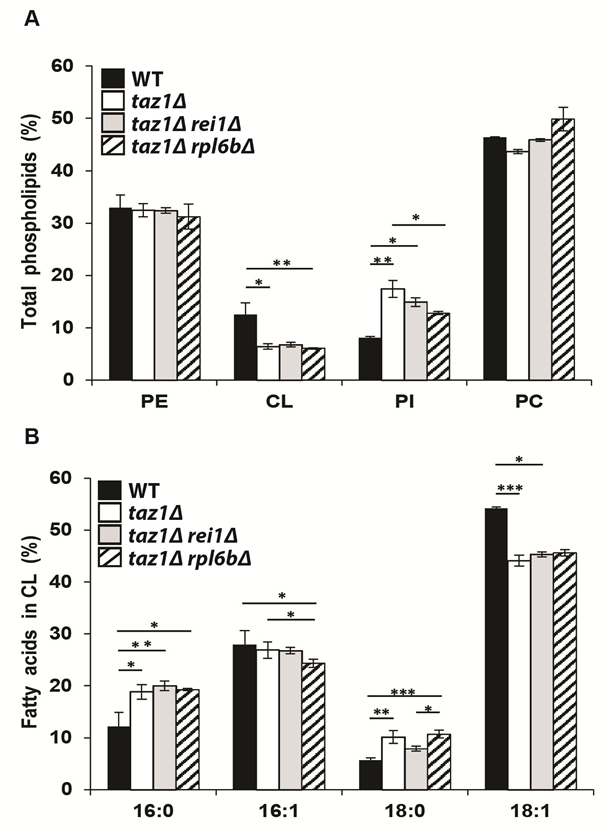 | FIGURE 2: Genetic ablation of REI1 or RPL6B in taz1Δ yeast does not restore cardiolipin remodeling. Lipids were extracted from mitochondria isolated from WT (black bars), taz1Δ (open bars), taz1Δ rei1Δ (grey bars) and taz1Δ rpl6bΔ (striped bars) cells grown in CSM 0.5% galactose + 2% ethanol at 36°C until a density of 2-3 OD600nm. (A) Relative contents of PE (phosphatidylethanolamine), CL (cardiolipin), PI (phosphatidylinositol) and PC (phosphatidylcholine) within each strain. (B) Relative fatty acid chain composition of CL within each strain (16:0, palmitic acid; 16:1, palmitoleic acid; 18:0, stearic acid; 18:1: oleic acid). Statistical analysis was done with Kruskal-Wallis test followed by Dunn’s multiple comparison test (*P<0.05; **P<0.01; ***P<0.001). Data are expressed as mean ± s.d. (n=4). The data for WT and taz1Δ strains were reported previously [60]. |
Partially decreasing cytosolic protein synthesis fully restores OXPHOS in taz1Δ yeast
As we have shown [60], the reduced ability of taz1Δ yeast to grow at 36°C in 2% ethanol + 0.5% galactose (shown in Fig. 1B) correlated with a decreased rate of oxygen con-sumption and diminished levels of key components involved in the transfer of electrons to oxygen, including complexes II-IV and cytochrome c. Accumulation of these proteins (Fig. 3A, B) and oxygen consumption measured with either NADH or Ascorbate-TMPD as electron donors were substantially improved after eliminating REI1 or RPL6B (Fig. 3C) or growing taz1Δ yeast in the presence of 10 nM CHX (Fig. 3D). Consequently, the rate of mitochondrial ATP synthesis was restored to almost normal levels (Fig. 3E).
–
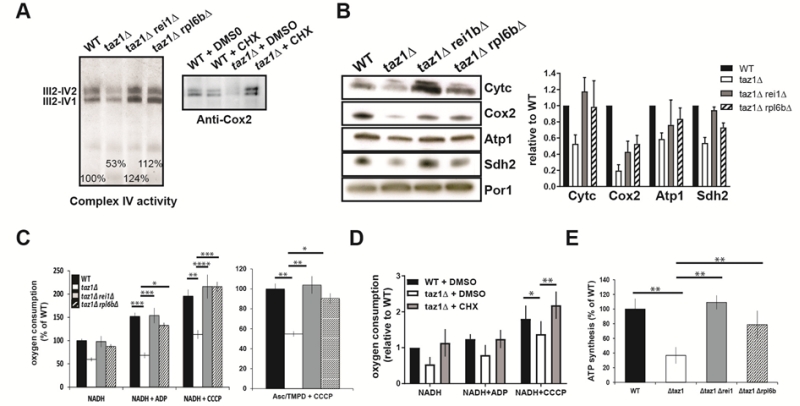 |
FIGURE 3: Partially decreasing cytosolic translation preserves oxidative phosphorylation in taz1Δ yeast. The experiments here described were performed using mitochondria isolated from cells grown for 24 hours at 36°C in CSM containing 0.5% galactose + 2% ethanol, supple-mented or not as indicated with 10 nM CHX, until a density of 2-3 OD600nm. (A, B) Steady-state levels of proteins involved in the transfer of electrons to oxygen. (A) Proteins were ex-tracted from the mitochondrial samples using 2 g digitonin per g of proteins. The supercomplexes III2-IV2 and III2-IV1 were revealed by the complex IV activity after separation by CN-PAGE or by western blot with antibodies against Cox2 in BN-PAGE gels. (B) Left panel. Total mitochondrial protein samples were resolved by SDS-PAGE (50 μg per lane) and probed with antibodies against the in-dicated proteins. The shown gels are representative of at least 3 experiments. Right panel. Quantification using ImageJ software. Levels of Cytc, Cox2, Atp1 and Sdh2 are normalized to Por1p and expressed relative to WT. (C) Genetic ablation of REI1 or RPL6B in taz1Δ yeast preserves mitochondrial respiration. On the left are the rates of oxygen consumption from NADH (4 mM) alone (state 4), after further addition (150 μM) of ADP (state 3) or CCCP (4 μM) (uncoupled respiration). The data are expressed in % of WT state 4 respiration (mean ± s.d, n=4). On the right are the oxygen consumption rates from electrons delivered directly to complex IV by ascorbate 12.5 mM/TMPD 1.4 mM in the presence of CCCP. Data are expressed relative to the WT (mean ± s.d, n=4). (D) Mitochondrial respiration is preserved in taz1Δ yeast grown in the presence of 10 nM CHX. NADH was used as the electron donor, as described in panel C (n=4). (E) ATP synthesis was measured using NADH as a respiratory substrate in the presence of 1 mM ADP. Data are expressed as mean ± s.d. (n=4) relative to the WT. The data for WT and taz1Δ strains were reported previously [60]. |
These observations were corroborated by monitoring changes in mitochondrial membrane potential (ΔΨ) with Rhodamine 123. As we showed [60], adding ADP to respiring taz1Δ mitochondria did not decrease ΔΨ due to their poor capacity to synthesize ATP. Consistent with their good ability to produce ATP, those from strains taz1Δ rei1Δ and taz1Δ rpl6bΔ efficiently responded to ADP as WT mitochondria (Fig. 4). Furthermore, after a subsequent addition of KCN, to block the respiratory chain, ΔΨ collapsed in one single rapid phase in taz1Δ mitochondria, whereas a residual potential was preserved in those from taz1Δ rei1Δ and taz1Δ rpl6bΔ and WT yeasts. This residual potential is due to the pumping of protons by ATP synthase, coupled by the hydrolysis of the ATP that accumulated in the mitochondrial matrix during phosphorylation of the added ADP, as evidenced by the loss of this potential by inhibiting ATP synthase with oligomycin (Fig. 4).
–
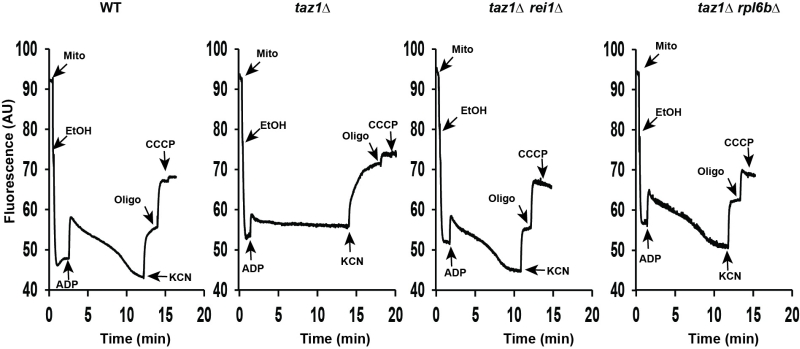 | FIGURE 4: Mitochondrial membrane potential. Variations in mitochondrial ΔΨ were moni-tored by fluorescence quenching of Rhodamine 123, using intact, osmotically-protected, mitochondria isolated from WT, taz1Δ, taz1Δ rei1Δ and taz1Δ rpl6bΔ cells grown in CSM containing 0.5% galactose + 2% ethanol at 36°C until a density of 2-3 OD600nm. The additions were 75 μM ADP, 0.5 μg/ml Rhodamine 123, 75 μg/ml mitochondrial proteins (Mito), 10 μl ethanol (EtOH), 2 mM potassium cyanide (KCN), 4 μM CCCP (carbonyl cyanide-m-chlorophenyl hydrazone) and 4 μg/ml oligomycin (oligo). The shown tracings are representative of four experimental trials. The data for WT and taz1Δ strains were reported previously [60]. |
Taken together, these observations demonstrate that partially decreasing cytosolic translation preserves the biogenesis and activity of the OXPHOS system in CL remodeling deficient yeast.
–
Partially decreasing cytosolic protein synthesis suppresses the enhanced production of ROS in taz1Δ yeast
Defects in the mitochondrial respiratory chain often result in a higher production/accumulation of reactive oxygen species (ROS) owing to an enhanced diversion of electrons from their normal pathway to oxygen, which was observed in CL remodeling deficient cells [65]. Thus, it was expected that taz1Δ yeast should produce less ROS after deleting REI1 or RPL6B or during growth in the presence of 10 nM CHX, which was indeed observed (Fig. 5).
–
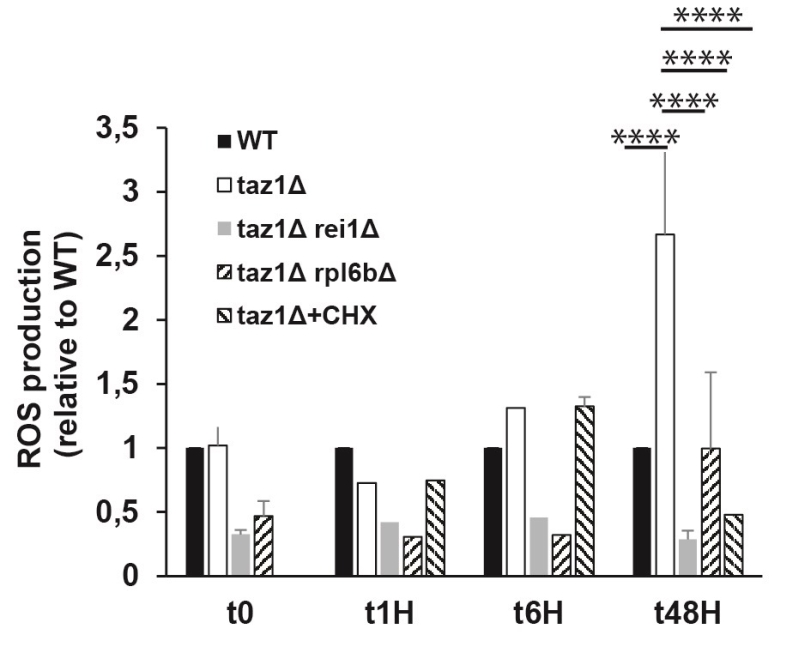 Impression | FIGURE 5: Partially decreasing cytosolic translation in taz1Δ yeast preserves a normal production of ROS. The cells were grown in CSM containing 0.5% galactose + 2% ethanol at 36°C for 48 hours. At the indicated times, ROS levels were measured by flow cytometry using dihydroethidium as a probe. The data are expressed in % relative to the WT at T0 (n=3). Statistical analysis was done with Tukey’s test (*P<0.05; **P<0.01; ***P<0.001; ****P<0.0001). The data for WT and taz1Δ strains were reported previously [60]. |
Decreasing cytosolic protein synthesis is also beneficial to tafazzin-deficient human cells
We aimed to understand whether partially decreasing cytosolic translation could also benefit human cells lacking Tafazzin. To this end, we used our previously described HeLa cells, in which the TAZ gene was knocked down by RNA interference (shTaz1) and two control cell lines, shWT1 and shTaz1R in which the expression of TAZ1 was not inhibited. As reported, ShTaz1 poorly accumulates Tafazzin, is defective in CL maturation, has a reduced capacity to associate respiratory chain complexes into ‘respirasomes’, produces abnormally enlarged cells and has a higher content in mitochondria compared to shWT1 and shTaz1R, as do cells from BTHS patients [24][48][66].
–
Herein we report that ShTaz1 cells proliferate four times slower and die more rapidly, in comparison to ShWT1 and ShTazR1 cells (Fig.6 A, B). We took advantage of these differences to test the capacity of CHX at counteracting the detrimental effects of a lack in CL maturation in human cells. A large beneficial effect was observed at a 50 pM concentration of CHX. In the experiment shown in Fig. 6C, the drug was added 24-25 hours after inoculating 200 µl of media with 5000 cells. CHX induced a 48 h lag phase, after which the cells grew for a long-lasting period of 170 hours, before dying and detaching from their support. In the absence of CHX, death was observed much more rapidly, after 72 hours of continuous growth. At the concentration of 50 pM, CHX had no effect on the proliferation of ShWT1 (Figure 6D). These observations indicate that a partial decrease in cytosolic translation is, as in taz1Δ yeast, beneficial to human cells defective in CL remodeling.
–
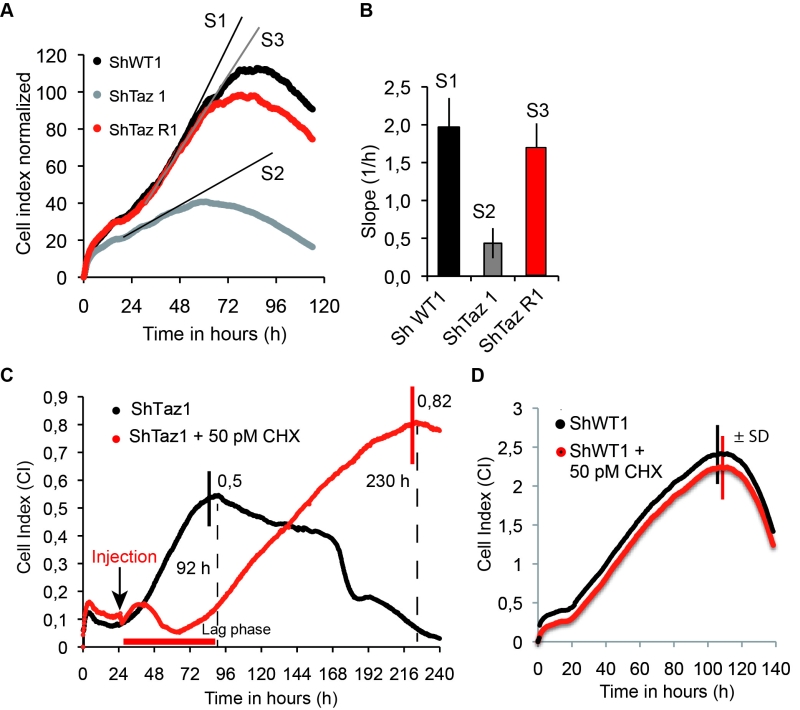 | FIGURE 6: CHX improves cell proliferation and viability of human Tafazzin-deficient cells. These experiments used our previously described HeLa cells in which TAZ gene has been knocked down by RNA interference (shTaz1) and two control cell lines, shWT1 and shTaz1R, in which expression of TAZ1 is not inhibited [67]. (A) Growth curves in 200 µL wells inoculated with 5 000 cells. After reaching a plateau, the cells die and detach from their support. S1, S2 and S3 are the slopes of the proliferation state for each cell lines. (B) Relative slopes (1/h) deduced from the proliferation curves shown in panel A. (C) xCELLigence recording of ShTaz1 proliferation in absence or presence of CHX at a concentration of 50 pM. CHX was added (black arrow) after a 24-hour adhesion step. The cultures were inoculated with 5 000 cells in 200 μL wells. The optimal Cell Index values are indicated along the curves as well as the time (in hours) taken to reach the inflexion point. The horizontal red bar stands for the 48 hours lag phase induced by CHX. Four independent experiments with 4 wells for each growth condition have been done (16 wells in total for establishing mean values). the ±SD is directly drawn on the top of the curves (in black for ShTaz1 and in red for SgTaz1 + 50 pM CHX). (D) xCELLigence recording of ShWT1 proliferation in absence or presence of CHX at a concentration of 50 pM. The cultures were inoculated with 5 000 cells in 200 μL wells. CHX was added before the binding of the cell to the substrate in this case in order to avoid the perturbations induced by the injection of CHX along the trace; the ±SD is directly drawn on the top of the curves (in black for ShWT1 and in red for SgTaz1, there is no significant variation). |
DISCUSSION
While a general inhibition of cytosolic protein synthesis would obviously be detrimental to the cell, our study reveals that a partial (50%) decrease in this activity preserves mtDNA maintenance and the biogenesis and activity of the oxidative phosphorylation (OXPHOS) system in a yeast model of the Barth syndrome, a mitochondrial disease associated to defects in the remodeling of cardiolipin (CL). The decreased mtDNA stability in taz1Δ yeast occurred in fermenting (glucose) cultures where the presence of this DNA is not indispensable. With a respiratory carbon source (glycerol), taz1Δ yeast cells lacking functional mtDNA were much less abundant owing to their incapacity to proliferate in these conditions. Thus, the respiratory deficiency of taz1Δ yeast does not result from a lack in mtDNA.
–
Importantly, CL remodeling was still deficient in taz1Δ cells rescued by a partial inhibition of cytosolic translation. This finding has two important corollaries: (i) the CL species remaining in the mutant (50% vs the WT) are sufficient for a proper biogenesis and functioning of the OXPHOS system, and (ii) the OXPHOS deficit is secondary to some other cellular dysfunction(s) that can be suppressed by decreasing cytosolic translation. At low concentration (50 pM), CHX also improved proliferation of HeLa cells deficient in tafazzin whereas that of WT Hela cells was not modified, which clearly demonstrates that growth improvement resulted from a compensation of the lack in tafazzin. Thus yeast and human CL remodeling deficient cells face similar difficulties that can be attenuated by targeting cytosolic translation. These finding reveal that a diminished capacity of CL remodeling deficient cells to preserve protein homeostasis is likely an important factor contributing to the pathogenesis of the Barth syndrome. This in turn, identifies cytosolic translation as a potential therapeutic target for the treatment of this disease.
–
Previous work revealed that decreasing cytosolic translation can also rescue yeast models of adPEO (autosomal dominant progressive external ophthalmoplegia) caused by mutations in ANT, a protein that exchanges adenine nucleotides across the mitochondrial inner membrane [67]. In addition to a defective exchange of adenine nucleotides, these mutations compromise the impermeability to protons of the inner membrane. Consequently, a sufficient electrical potential cannot be maintained across this mem-brane and this impedes many proteins to reach the organelle. This has deleterious consequences, not only for mitochondria, but also for the cytosol that is then confronted with the over-accumulation and the misfolding of mitochondrial precursor proteins [63]. This protein stress, which was termed mitochondrial precursor over-accumulation stress (mPOS), was shown to induce a cellular response, named unfolded protein response activated by mistargeted mitochondrial proteins (UPRam), that is mainly characterized by a decreased rate of cytosolic protein synthesis and a faster rate of protein clearance by the proteasome [67][68]. Consistent with these findings, mutations in proteins directly involved in the transport of proteins into mitochondria were found to similarly poison the cytosol with mitochondrial proteins triggering the cell to diminish the production and stimulate the degradation of proteins [68]. The significant drop (35%) in cytosolic translation in taz1Δ yeast is an indication that a lack in CL remodeling could as well lead to the congestion of the cytosol with miss-localized mitochondrial proteins.
–
Considering the numerous roles of CL in mitochondria, it might be that a lack in the remodeling of this phospholipid affects the capacity of mitochondria to import proteins, and thereby makes other compartments of the cell more susceptible to protein stress. In support to this hypothesis, loss of CL remodeling was shown to partially compromise the biogenesis of the protein translocase (TOM) and the sorting and assembly machinery (SAM) of the outer mitochondrial membrane [18][69]. In another study [36], no evidence was found for a decreased preprotein accumulation in vitro with taz1-deficient mitochondria. However, because these assays used minute amounts of preproteins it might be difficult to detect in this way a partially diminished protein import capacity.
–
The recovery of mitochondrial function in taz1Δ yeast by a partial decrease of cytosolic translation is in line with a recent study showing that CL has an important role in promoting the induction of a mitochondrial-to-cytosolic stress response (MCSR) that enables the cell to improve protein homeostasis in both compartments [70]. Consistently also, it was shown that rapamycin, a specific inhibitor of the mTOR signaling pathway that regulates several extra-mitochondrial cellular pathways among which protein synthesis, robustly enhances survival and attenuates the disease’s progression in a mouse model and patient cells of the Leigh Syndrome, one of the most devastating mitochondrial disorders [71][72].
–
Clearly, beyond a certain level of mitochondrial damage, the protein stress responsive pathways may not be sufficient. This may explain that in addition to its spontaneous 35% drop in protein synthesis, taz1Δ yeast requires a further (15%) decrease in this activity to be effectively rescued. Whether a general protein synthesis inhibition or a reduced production of specific proteins is beneficial to CL remodeling deficient cells is an interesting and important issue. In this respect, it is interesting to note that Gerst et al. found that ribosomal protein paralogs specifically modulate translation of mitochondrial precursor proteins [73]. Such observations hold promise for the development of more targeted therapeutic approaches with less undesirable side-effects to preserve protein homeostasis in cells poisoned by the over-accumulation in the cytosol of mitochondrial protein precursors.
MATERIALS AND METHODS
Growth media
The following media were used for growing yeast: Fermentable YPAD media containing 1% (w/v) yeast extract, 1% (w/v) bacto peptone, 40 mg/L adenine and 2% (v/v) glucose; Respiratory rich media YPAEthanol containing 1% (w/v) yeast extract, 1% (w/v) bacto peptone, 40 mg/L adenine and 2% (v/v) ethanol; Non-fermentable complete synthetic media CSM/gal/ethanol containing 0.17% (w/v) yeast nitrogen base without aminoacids and ammonium sulfate, 0.5% (w/v) ammonium sulfate, 0.5% (w/v) galactose, 2% ethanol and 0.08% (w/v) of a mixture of aminoacids and bases from Formedium. Solid media contained 2% (w/v) agar.
–
Construction of strains taz1Δ rpl6bΔ and taz1Δ rei1Δ
These strains were constructed by deleting entirely the open reading frame of RPL6B or REI1 with the KanMX marker using a described procedure [74] in taz1Δ mutant (MATa ade2-1 ura3-1 his3-11, 15 trp1-1 leu2-3,112 can1-100, taz1::TRP1). For the construction of the strain taz1Δ rpl6bΔ, the KanMX cassette was amplified using primers pFA6a-Kan: Rpl6b-del-F (CTT TCT TGA ACT TGG AAG AGA AGC AAA TAT ATT CAA CGA A cgg atc ccc ggg tta att aa) and Rpl6b-del-R (CTA TTT TAA ATC ATT TAT AAT TTT TTC AGT TCA AT gaa ttc gag ctc gtt taa ac) (the sequences in capital letters are homologous to the RPL6B flanking regions, those in lower case enable amplication of KanMX. For the construction of the strain taz1Δ rei1Δ, the KanMX cassette was amplified with pFA6a-Kan: Rei1-del-F (CAT TAG AAG TCA AGA AGA GAG CAT ATC AGT AAC AAT ACG cgg atc ccc ggg tta attaa) and Rei1-del-R (GCG ACA AAA TAC TAA AAA AAG TAG TGC AAA AAG AA gaa ttc gag ctc gtt taa ac). The primers Rpl6b-Fbis (CTG CGC TTC CGT TCA GCA TC), Rpl6b-Rbis (CGA TGA CCT GAT CTT GAA CCC) or, Rei1-Fbis (GTG GTG TAG CTA TTT GTA CAT G), and Rei1-Rbis (CAA CAT CTT CAG TCT TCA GCA GC) were used to verify the deletions of REI1 and RPL6B by PCR.
–
Yeast-based drug assay
0.125 OD of exponentially growing cell were homogeneously spread with sterile glass beads on a square Petri dish (12 cm x 12cm) containing solid YPA ethanol medium. Sterile filters were deposited on the plate and spotted with cycloheximide (purchased from Sigma), anisomycin (purchased from Sigma), and emetine (purchased from Sigma) dissolved in DMSO. Growth improvement was assessed after several days of incubation at 36°C.
–
Bioenergetics experiments
The mitochondria were prepared by the enzymatic method as described [75]. Protein concentrations were determined by the Lowry method [76] in the presence of 5% SDS. Oxygen consumption rates were measured on 75 μg of fresh mitochondria using a Clarke electrode in the respiration buffer (0.65 M mannitol, 0.36 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic, 5 mM tris-phosphate, 10 mM tris-maleate, pH 6.8) as described [77] (see legend of Fig. 3 for the concentrations of reagents used). Variations in mitochondrial transmembrane potential (ΔΨ) were evaluated in the same respiration buffer, by monitoring the quenching of rhodamine 123 fluorescence (0.5 mg/ml) using a λexc of 485 nm and a λem of 525 nm under constant stirring, using a FLX Spectrofluorimeter (SAFAS, Monaco), as described [78]. ATP synthesis rates were measured using 75 μg of fresh mitochondria in a 2-ml thermostatically controlled chamber at 28°C in respiration buffer, in the presence of 4 mM NADH and 1mM ADP as described [79]. Aliquots were withdrawn from the oxygraph cuvette every 15 seconds and supplemented with 2.5% (w/v) perchloric acid, 8.5 mM EDTA to stop the reaction and then neutralized to pH 6.8 by adding 2N KOH, 0.3 M MOPS. ATP was quantified using a luciferin/luciferase assay (ATPLite kit from Perkin Elmer) on a LKB bioluminometer. The participation of the F1F0-ATP synthase in ATP production was assessed using the same protocol, in the presence of oligomycin (3 µg/ml).
–
BN/CN-PAGE & SDS-PAGE
Blue native BN-PAGE and clear native CN-PAGE experiments were carried out as described [80], using mitochondrial extracts solubilized with digitonin (2 gr per gr protein) run in 3–12% continuous gradient acrylamide gels. The in-gel complex IV activity was revealed using a solution of Tris 5mM pH 7.4, diaminobenzidine 0.5 mg/ml, cytochrome c 0.05 mM. The proteins were also analyzed by Western blotting on poly(vinylidene difluoride) membranes as described [81]. Polyclonal antibodies raised against yeast ATP synthase were used at a dilution of 1:50000 for subunit α (gift from J. Velours, IBGC, Bordeaux, France); 1:10000 for cytochrome c (gift from S. Manon, IBGC, Bordeaux, France); 1:5000 for succinate dehydrogenase Sdh2 subunit (gift from C. Dallabona, University of Parma, Italy). Monoclonal antibodies against porin and Cox2 (from Molecular Probes) were used at a dilution of 1:5000. Nitrocellulose membranes were incubated with peroxidase-labeled antibodies (from Promega) at a 1:2500 dilution, and analyzed by electrochemiluminescence. Quantification of the protein signals was performed with the ImageJ software.
–
In vivo labeling of mitochondrial translation products
The indicated strains were grown to early exponential phase (OD/ml of 2) in 20 ml of rich ethanol/galactose media at 36°C. The cells were harvested by centrifugation and washed twice with a minimum medium containing 2% ethanol and 0,5% galactose, supplemented with histidine, tryptophan, leucine, uracil and adenine (50 mg/liter each). To evaluate total protein synthesis, cells were resuspended in 1 ml of the same medium with the addition of 55 µCi of [35S] methionine plus [35S] cysteine (Amersham Biosciences) and incubated for 20 min at 36°C. To evaluate mitochondrial protein synthesis the same procedure was followed but before adding [35S] methionine plus [35S] cysteine, cells were first treated with 7,5 mg/ml cycloheximide during 5 minutes. After the labeling reactions, total protein extracts were prepared and quantified using the Lowry method. The proteins were separated by SDS-PAGE on a 12% polyacrylamide gel, transferred onto a nitrocellulose membrane and analyzed with a PhosphorImager. Quantification of radioactive proteins was performed using the software ImageJ [82].
–
Lipid analyses
Mitochondrial lipids were analyzed as described [60]. In summary, the lipids were extracted with 2 ml of chloroform/methanol (2:1, v/v). After centrifugation, the organic phase was isolated and the remaining lipids were further extracted twice by adding 2 ml of chloroform to the aqueous phase. The organic phases were pooled and evaporated to dryness. The lipids were then resuspended in chloroform/methanol (2:1, v/v). Respective volumes equivalent to 50 µg of acyl chains were spotted on silica plates, four times for each strain. Polar lipids were separated by one dimensional TLC using chloroform/methanol/1-propanol/methyl acetate/0.25% KCl (10:4:10:10:3.6, by vol.) as a solvent [83]. The lipids were located by immersing the plates in a solution of 0.001% (w/v) primuline in PBS, followed by visualization under UV light. The zones of the gel corresponding to PE, CL, PI and PC were then scraped and added to 1 ml of methanol/2.5% H2SO4 containing 5 μg of heptadecanoic acid methyl ester as a standard. The lipid mixtures were incubated at 80◦C for 1 h, and 1.5 ml of water and 400 µl of hexane were then added. After centrifugation, the hexane phase containing FAMES (fatty acid methyl esters) was isolated. Separation of FAMES was performed as described [39].
–
ROS analysis.
Cells at 0.4 OD units were taken from liquid cultures, pelleted in a microcentrifuge, resuspended in 1 ml of phosphate-buffered saline (PBS) containing 50 μM dihydroethidium (DHE; Molecular Probes) and incubated at room temperature for 5 min. Flow cytometry was carried out on a Becton-Dickinson Accuri C6 model flow cytometer. The DHE fluorescence indicated was the direct output of the FL2A (red fluorescence) channel without compensation. A total of 100,000 cells were analyzed for each curve.
–
HeLa cells culture and transfection
The cervical carcinoma HeLa cell lines were cultured in DME supplemented with 10% FCS and L- glutamine. Transfection of HeLa cells was performed using Lipofectamine 2000 (Invitrogen). Bcl-xL, shTaz, and shCont stable HeLa cell lines were generated by transfection with pcDNA3/Bcl-xL, pSUPER/shTaz, or pSUPER/shCont, respectively, and selected in G418 [84]. The revertant shTaz1R cell line was generated by cotransfecting shTaz1 HeLa cells with pLpC vector (carrying a puromycin resistance gene), and pcDNA3/Taz mut and stable clones were selected in the presence of puromycin [84].
–
xCELLigence real time cellular proliferation measurements
Experiments were carried out using the xCELLigence RTCA DP instrument (ACEA Biosciences, Ozyme, France) placed in a humidified incubator at 37°C and 5% CO2. Cell proliferation and cytotoxicity experiments were performed using 16-well plates (E-plate, Ozyme, Montigny le Bretonneux, France). The microelectrodes attached at the bottom of the wells allowed for impedance-based detection of the attachment, spreading and proliferation of the cells. Initially, 180 μL of cell-free growth medium (10% FBS) was added to the wells. After leaving the devices at room temperature for 30 min, the background impedance for each well was measured. Cells were harvested from exponential phase cultures by a standardized detachment procedure using 0.05% Trypsin-EDTA (Invitrogen). Flow cytometry was used to count the cells and test their viability (FSC versus propidium iodide staining). 5000 or 7500 cells in 20 μl were added in each well. After leaving the plates at room temperature for 30 min to allow early cell attachment, in accordance with the manufacturer’s guidelines, they were locked in the RTCA DP device in the incubator and the impedance value of each well was automatically monitored by the xCELLigence system and expressed as a Cell Index value (CI). Water was added to the space surrounding the wells of the E-plate to avoid interference from evaporation. For proliferation assays, the cells were incubated during 120h for toxicity in growth medium (10% FBS) and CI was monitored every 15 min during the whole duration of the experiment. Four replicates of each conditions were used in each test. After an initial assessment of the concentration of cycloheximide which would be non-toxic to the ShWT1 cells, we used a test of proliferation in presence and absence of low doses of cycloheximide (1 pM to 100 pM) either prior to cell seeding or after an initial adhesion phase and at an early proliferative step (at 24-26 hours). All experiments were conducted over 240 hours. All plots were normalized to the Cell Index.
References
- P. Barth, H. Scholte, J. Berden, J. Van Der Klei-Van Moorsel, I. Luyt-Houwen, E. Van'T Veer-Korthof, J. Van Der Harten, and M. Sobotka-Plojhar, "An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes", Journal of the Neurological Sciences, vol. 62, pp. 327-355, 1983. http://dx.doi.org/10.1016/0022-510X(83)90209-5
- P.G. Barth, F. Valianpour, V.M. Bowen, J. Lam, M. Duran, F.M. Vaz, and R.J. Wanders, "X‐linked cardioskeletal myopathy and neutropenia (Barth syndrome): An update", American Journal of Medical Genetics Part A, vol. 126A, pp. 349-354, 2004. http://dx.doi.org/10.1002/ajmg.a.20660
- P.A. Bolhuis, G.W. Hensels, T.J. Hulsebos, F. Baas, and P.G. Barth, "Mapping of the locus for X-linked cardioskeletal myopathy with neutropenia and abnormal mitochondria (Barth syndrome) to Xq28.", American journal of human genetics, 1991. http://www.ncbi.nlm.nih.gov/pubmed/1998334
- F.L. Hoch, "Cardiolipins and biomembrane function", Biochimica et Biophysica Acta (BBA) - Reviews on Biomembranes, vol. 1113, pp. 71-133, 1992. http://dx.doi.org/10.1016/0304-4157(92)90035-9
- M. Schlame, and D. Haldar, "Cardiolipin is synthesized on the matrix side of the inner membrane in rat liver mitochondria.", The Journal of biological chemistry, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8380172
- M. Schlame, D. Rua, and M.L. Greenberg, "The biosynthesis and functional role of cardiolipin", Progress in Lipid Research, vol. 39, pp. 257-288, 2000. http://dx.doi.org/10.1016/S0163-7827(00)00005-9
- N. Ikon, B. Su, F. Hsu, T.M. Forte, and R.O. Ryan, "Exogenous cardiolipin localizes to mitochondria and prevents TAZ knockdown-induced apoptosis in myeloid progenitor cells", Biochemical and Biophysical Research Communications, vol. 464, pp. 580-585, 2015. http://dx.doi.org/10.1016/j.bbrc.2015.07.012
- R. Bligny, and R. Douce, "A precise localization of cardiolipin in plant cells", Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism, vol. 617, pp. 254-263, 1980. http://dx.doi.org/10.1016/0005-2760(80)90168-X
- K. Hostetler, H. Van Den Bosch, and L. Van Deenen, "The mechanism of cardiolipin biosynthesis in liver mitochondria", Biochimica et Biophysica Acta (BBA) - Lipids and Lipid Metabolism, vol. 260, pp. 507-513, 1972. http://dx.doi.org/10.1016/0005-2760(72)90065-3
- A.S. Joshi, J. Zhou, V.M. Gohil, S. Chen, and M.L. Greenberg, "Cellular functions of cardiolipin in yeast", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1793, pp. 212-218, 2009. http://dx.doi.org/10.1016/j.bbamcr.2008.07.024
- S. Bazán, E. Mileykovskaya, V.K.P. Mallampalli, P. Heacock, G.C. Sparagna, and W. Dowhan, "Cardiolipin-dependent Reconstitution of Respiratory Supercomplexes from Purified Saccharomyces cerevisiae Complexes III and IV", Journal of Biological Chemistry, vol. 288, pp. 401-411, 2013. http://dx.doi.org/10.1074/jbc.M112.425876
- K. Pfeiffer, V. Gohil, R.A. Stuart, C. Hunte, U. Brandt, M.L. Greenberg, and H. Schägger, "Cardiolipin Stabilizes Respiratory Chain Supercomplexes", Journal of Biological Chemistry, vol. 278, pp. 52873-52880, 2003. http://dx.doi.org/10.1074/jbc.M308366200
- M. Zhang, E. Mileykovskaya, and W. Dowhan, "Gluing the Respiratory Chain Together", Journal of Biological Chemistry, vol. 277, pp. 43553-43556, 2002. http://dx.doi.org/10.1074/jbc.C200551200
- A.S. Joshi, M.N. Thompson, N. Fei, M. Hüttemann, and M.L. Greenberg, "Cardiolipin and Mitochondrial Phosphatidylethanolamine Have Overlapping Functions in Mitochondrial Fusion in Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 287, pp. 17589-17597, 2012. http://dx.doi.org/10.1074/jbc.M111.330167
- T. Ban, J.A. Heymann, Z. Song, J.E. Hinshaw, and D.C. Chan, "OPA1 disease alleles causing dominant optic atrophy have defects in cardiolipin-stimulated GTP hydrolysis and membrane tubulation", Human Molecular Genetics, vol. 19, pp. 2113-2122, 2010. http://dx.doi.org/10.1093/hmg/ddq088
- R.M. DeVay, L. Dominguez-Ramirez, L.L. Lackner, S. Hoppins, H. Stahlberg, and J. Nunnari, "Coassembly of Mgm1 isoforms requires cardiolipin and mediates mitochondrial inner membrane fusion", Journal of Cell Biology, vol. 186, pp. 793-803, 2009. http://dx.doi.org/10.1083/jcb.200906098
- F. Jiang, M.T. Ryan, M. Schlame, M. Zhao, Z. Gu, M. Klingenberg, N. Pfanner, and M.L. Greenberg, "Absence of Cardiolipin in the crd1 Null Mutant Results in Decreased Mitochondrial Membrane Potential and Reduced Mitochondrial Function", Journal of Biological Chemistry, vol. 275, pp. 22387-22394, 2000. http://dx.doi.org/10.1074/jbc.M909868199
- N. Gebert, A.S. Joshi, S. Kutik, T. Becker, M. McKenzie, X.L. Guan, V.P. Mooga, D.A. Stroud, G. Kulkarni, M.R. Wenk, P. Rehling, C. Meisinger, M.T. Ryan, N. Wiedemann, M.L. Greenberg, and N. Pfanner, "Mitochondrial Cardiolipin Involved in Outer-Membrane Protein Biogenesis: Implications for Barth Syndrome", Current Biology, vol. 19, pp. 2133-2139, 2009. http://dx.doi.org/10.1016/j.cub.2009.10.074
- V.A. Patil, J.L. Fox, V.M. Gohil, D.R. Winge, and M.L. Greenberg, "Loss of Cardiolipin Leads to Perturbation of Mitochondrial and Cellular Iron Homeostasis", Journal of Biological Chemistry, vol. 288, pp. 1696-1705, 2013. http://dx.doi.org/10.1074/jbc.M112.428938
- C.T. Chu, H. Bayır, and V.E. Kagan, "LC3 binds externalized cardiolipin on injured mitochondria to signal mitophagy in neurons", Autophagy, vol. 10, pp. 376-378, 2013. http://dx.doi.org/10.4161/auto.27191
- C.T. Chu, J. Ji, R.K. Dagda, J.F. Jiang, Y.Y. Tyurina, A.A. Kapralov, V.A. Tyurin, N. Yanamala, I.H. Shrivastava, D. Mohammadyani, K.Z. Qiang Wang, J. Zhu, J. Klein-Seetharaman, K. Balasubramanian, A.A. Amoscato, G. Borisenko, Z. Huang, A.M. Gusdon, A. Cheikhi, E.K. Steer, R. Wang, C. Baty, S. Watkins, I. Bahar, H. Bayır, and V.E. Kagan, "Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells", Nature Cell Biology, vol. 15, pp. 1197-1205, 2013. http://dx.doi.org/10.1038/ncb2837
- P. Hsu, X. Liu, J. Zhang, H. Wang, J. Ye, and Y. Shi, "Cardiolipin remodeling by TAZ/tafazzin is selectively required for the initiation of mitophagy", Autophagy, vol. 11, pp. 643-652, 2015. http://dx.doi.org/10.1080/15548627.2015.1023984
- X. Li, B. Tsoi, Y. Li, H. Kurihara, and R. He, "Cardiolipin and Its Different Properties in Mitophagy and Apoptosis", Journal of Histochemistry & Cytochemistry, vol. 63, pp. 301-311, 2015. http://dx.doi.org/10.1369/0022155415574818
- F. Gonzalvez, M. D'Aurelio, M. Boutant, A. Moustapha, J. Puech, T. Landes, L. Arnauné-Pelloquin, G. Vial, N. Taleux, C. Slomianny, R.J. Wanders, R.H. Houtkooper, P. Bellenguer, I.M. Møller, E. Gottlieb, F.M. Vaz, G. Manfredi, and P.X. Petit, "Barth syndrome: Cellular compensation of mitochondrial dysfunction and apoptosis inhibition due to changes in cardiolipin remodeling linked to tafazzin (TAZ) gene mutation", Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, vol. 1832, pp. 1194-1206, 2013. http://dx.doi.org/10.1016/j.bbadis.2013.03.005
- B. Heit, T. Yeung, and S. Grinstein, "Changes in mitochondrial surface charge mediate recruitment of signaling molecules during apoptosis", American Journal of Physiology-Cell Physiology, vol. 300, pp. C33-C41, 2011. http://dx.doi.org/10.1152/ajpcell.00139.2010
- T. Kim, Y. Zhao, W. Ding, J.N. Shin, X. He, Y. Seo, J. Chen, H. Rabinowich, A.A. Amoscato, and X. Yin, "Bid-Cardiolipin Interaction at Mitochondrial Contact Site Contributes to Mitochondrial Cristae Reorganization and CytochromecRelease", Molecular Biology of the Cell, vol. 15, pp. 3061-3072, 2004. http://dx.doi.org/10.1091/mbc.E03-12-0864
- V. Manganelli, A. Capozzi, S. Recalchi, M. Signore, V. Mattei, T. Garofalo, R. Misasi, M. Degli Esposti, and M. Sorice, "Altered Traffic of Cardiolipin during Apoptosis: Exposure on the Cell Surface as a Trigger for “Antiphospholipid Antibodies”", Journal of Immunology Research, vol. 2015, pp. 1-9, 2015. http://dx.doi.org/10.1155/2015/847985
- J.B. McMillin, and W. Dowhan, "Cardiolipin and apoptosis", Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, vol. 1585, pp. 97-107, 2002. http://dx.doi.org/10.1016/S1388-1981(02)00329-3
- V. KOSHKIN, and M.L. GREENBERG, "Oxidative phosphorylation in cardiolipin-lacking yeast mitochondria", Biochemical Journal, vol. 347, pp. 687-691, 2000. http://dx.doi.org/10.1042/bj3470687
- V. KOSHKIN, and M.L. GREENBERG, "Cardiolipin prevents rate-dependent uncoupling and provides osmotic stability in yeast mitochondria", Biochemical Journal, vol. 364, pp. 317-322, 2002. http://dx.doi.org/10.1042/bj3640317
- B. Kadenbach, P. Mende, H. Kolbe, I. Stipani, and F. Palmieri, "The mitochondrial phosphate carrier has an essential requirement for cardiolipin", FEBS Letters, vol. 139, pp. 109-112, 1982. http://dx.doi.org/10.1016/0014-5793(82)80498-5
- N.C. Robinson, "Functional binding of cardiolipin to cytochromec oxidase", Journal of Bioenergetics and Biomembranes, vol. 25, pp. 153-163, 1993. http://dx.doi.org/10.1007/bf00762857
- H. NOËL, and S.V. PANDE, "An essential requirement of cardiolipin for mitochondrial carnitine acylcarnitine translocase activity", European Journal of Biochemistry, vol. 155, pp. 99-102, 1986. http://dx.doi.org/10.1111/j.1432-1033.1986.tb09463.x
- F.M. Vaz, R.H. Houtkooper, F. Valianpour, P.G. Barth, and R.J. Wanders, "Only One Splice Variant of the Human TAZ Gene Encodes a Functional Protein with a Role in Cardiolipin Metabolism", Journal of Biological Chemistry, vol. 278, pp. 43089-43094, 2003. http://dx.doi.org/10.1074/jbc.M305956200
- Z. Gu, F. Valianpour, S. Chen, F.M. Vaz, G.A. Hakkaart, R.J.A. Wanders, and M.L. Greenberg, "Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome", Molecular Microbiology, vol. 51, pp. 149-158, 2003. http://dx.doi.org/10.1046/j.1365-2958.2003.03802.x
- K. Brandner, D.U. Mick, A.E. Frazier, R.D. Taylor, C. Meisinger, and P. Rehling, "Taz1, an Outer Mitochondrial Membrane Protein, Affects Stability and Assembly of Inner Membrane Protein Complexes: Implications for Barth Syndrome", Molecular Biology of the Cell, vol. 16, pp. 5202-5214, 2005. http://dx.doi.org/10.1091/mbc.E05-03-0256
- A.F. Neuwald, "Barth syndrome may be due to an acyltransferase deficiency", Current Biology, vol. 7, pp. R462-R466, 1997. http://dx.doi.org/10.1016/s0960-9822(06)00237-5
- Y. Xu, R.I. Kelley, T.J. Blanck, and M. Schlame, "Remodeling of Cardiolipin by Phospholipid Transacylation", Journal of Biological Chemistry, vol. 278, pp. 51380-51385, 2003. http://dx.doi.org/10.1074/jbc.M307382200
- E. TESTET, J. LAROCHE-TRAINEAU, A. NOUBHANI, D. COULON, O. BUNOUST, N. CAMOUGRAND, S. MANON, R. LESSIRE, and J. BESSOULE, "Ypr140wp, ‘the yeast tafazzin’, displays a mitochondrial lysophosphatidylcholine (lyso-PC) acyltransferase activity related to triacylglycerol and mitochondrial lipid synthesis", Biochemical Journal, vol. 387, pp. 617-626, 2005. http://dx.doi.org/10.1042/BJ20041491
- M. Ren, C.K. Phoon, and M. Schlame, "Metabolism and function of mitochondrial cardiolipin", Progress in Lipid Research, vol. 55, pp. 1-16, 2014. http://dx.doi.org/10.1016/j.plipres.2014.04.001
- A. Sickmann, J. Reinders, Y. Wagner, C. Joppich, R. Zahedi, H.E. Meyer, B. Schönfisch, I. Perschil, A. Chacinska, B. Guiard, P. Rehling, N. Pfanner, and C. Meisinger, "The proteome of Saccharomyces cerevisiae mitochondria", Proceedings of the National Academy of Sciences, vol. 100, pp. 13207-13212, 2003. http://dx.doi.org/10.1073/pnas.2135385100
- P. Vreken, F. Valianpour, L.G. Nijtmans, L.A. Grivell, B. Plecko, R.J. Wanders, and P.G. Barth, "Defective Remodeling of Cardiolipin and Phosphatidylglycerol in Barth Syndrome", Biochemical and Biophysical Research Communications, vol. 279, pp. 378-382, 2000. http://dx.doi.org/10.1006/bbrc.2000.3952
- M. Schlame, R.I. Kelley, A. Feigenbaum, J.A. Towbin, P.M. Heerdt, T. Schieble, R.J. Wanders, S. DiMauro, and T.J. Blanck, "Phospholipid abnormalities in children with Barth syndrome", Journal of the American College of Cardiology, vol. 42, pp. 1994-1999, 2003. http://dx.doi.org/10.1016/j.jacc.2003.06.015
- F. Valianpour, V. Mitsakos, D. Schlemmer, J.A. Towbin, J.M. Taylor, P.G. Ekert, D.R. Thorburn, A. Munnich, R.J. Wanders, P.G. Barth, and F.M. Vaz, "Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis", Journal of Lipid Research, vol. 46, pp. 1182-1195, 2005. http://dx.doi.org/10.1194/jlr.M500056-JLR200
- L. Ferri, M.A. Donati, S. Funghini, S. Malvagia, S. Catarzi, L. Lugli, L. Ragni, E. Bertini, F.M. Vaz, D.N. Cooper, R. Guerrini, and A. Morrone, "New clinical and molecular insights on Barth syndrome", Orphanet Journal of Rare Diseases, vol. 8, 2013. http://dx.doi.org/10.1186/1750-1172-8-27
- F. Gonzalvez, and E. Gottlieb, "Cardiolipin: Setting the beat of apoptosis", Apoptosis, vol. 12, pp. 877-885, 2007. http://dx.doi.org/10.1007/s10495-007-0718-8
- J. Dudek, I. Cheng, M. Balleininger, F.M. Vaz, K. Streckfuss-Bömeke, D. Hübscher, M. Vukotic, R.J. Wanders, P. Rehling, and K. Guan, "Cardiolipin deficiency affects respiratory chain function and organization in an induced pluripotent stem cell model of Barth syndrome", Stem Cell Research, vol. 11, pp. 806-819, 2013. http://dx.doi.org/10.1016/j.scr.2013.05.005
- Y. Xu, J.J. Sutachan, H. Plesken, R.I. Kelley, and M. Schlame, "Erratum: Characterization of lymphoblast mitochondria from patients with Barth syndrome", Laboratory Investigation, vol. 85, pp. 831, 2005. http://dx.doi.org/10.1038/labinvest.3700290
- L.M. Steinmetz, C. Scharfe, A.M. Deutschbauer, D. Mokranjac, Z.S. Herman, T. Jones, A.M. Chu, G. Giaever, H. Prokisch, P.J. Oefner, and R.W. Davis, "Systematic screen for human disease genes in yeast", Nature Genetics, vol. 31, pp. 400-404, 2002. http://dx.doi.org/10.1038/ng929
- J. Reinders, R.P. Zahedi, N. Pfanner, C. Meisinger, and A. Sickmann, "Toward the Complete Yeast Mitochondrial Proteome: Multidimensional Separation Techniques for Mitochondrial Proteomics", Journal of Proteome Research, vol. 5, pp. 1543-1554, 2006. http://dx.doi.org/10.1021/pr050477f
- H. Prokisch, C. Scharfe, D.G. Camp, W. Xiao, L. David, C. Andreoli, M.E. Monroe, R.J. Moore, M.A. Gritsenko, C. Kozany, K.K. Hixson, H.M. Mottaz, H. Zischka, M. Ueffing, Z.S. Herman, R.W. Davis, T. Meitinger, P.J. Oefner, R.D. Smith, and L.M. Steinmetz, "Integrative Analysis of the Mitochondrial Proteome in Yeast", PLoS Biology, vol. 2, pp. e160, 2004. http://dx.doi.org/10.1371/journal.pbio.0020160
- D.J. Pagliarini, S.E. Calvo, B. Chang, S.A. Sheth, S.B. Vafai, S. Ong, G.A. Walford, C. Sugiana, A. Boneh, W.K. Chen, D.E. Hill, M. Vidal, J.G. Evans, D.R. Thorburn, S.A. Carr, and V.K. Mootha, "A Mitochondrial Protein Compendium Elucidates Complex I Disease Biology", Cell, vol. 134, pp. 112-123, 2008. http://dx.doi.org/10.1016/j.cell.2008.06.016
- H. Rhee, P. Zou, N.D. Udeshi, J.D. Martell, V.K. Mootha, S.A. Carr, and A.Y. Ting, "Proteomic Mapping of Mitochondria in Living Cells via Spatially Restricted Enzymatic Tagging", Science, vol. 339, pp. 1328-1331, 2013. http://dx.doi.org/10.1126/science.1230593
- N. Bonnefoy, and T.D. Fox, "Genetic transformation of Saccharomyces cerevisiae mitochondria", Methods in Cell Biology, pp. 381-396, 2001. http://dx.doi.org/10.1016/s0091-679x(01)65022-2
- A. Tzagoloff, and C.L. Dieckmann, "PET genes of Saccharomyces cerevisiae.", Microbiological reviews, 1990. http://www.ncbi.nlm.nih.gov/pubmed/2215420
- S. Claypool, "The power of yeast to model diseases of the powerhouse of the cell", Frontiers in Bioscience, vol. 18, pp. 241, 2013. http://dx.doi.org/10.2741/4098
- J. Lasserre, A. Dautant, R.S. Aiyar, R. Kucharczyk, A. Glatigny, D. Tribouillard-Tanvier, J. Rytka, M. Blondel, N. Skoczen, P. Reynier, L. Pitayu, A. Rötig, A. Delahodde, L.M. Steinmetz, G. Dujardin, V. Procaccio, and J. di Rago, "Yeast as a system for modeling mitochondrial disease mechanisms and discovering therapies", Disease Models & Mechanisms, vol. 8, pp. 509-526, 2015. http://dx.doi.org/10.1242/dmm.020438
- E. Couplan, R.S. Aiyar, R. Kucharczyk, A. Kabala, N. Ezkurdia, J. Gagneur, R.P. St. Onge, B. Salin, F. Soubigou, M. Le Cann, L.M. Steinmetz, J. di Rago, and M. Blondel, "A yeast-based assay identifies drugs active against human mitochondrial disorders", Proceedings of the National Academy of Sciences, vol. 108, pp. 11989-11994, 2011. http://dx.doi.org/10.1073/pnas.1101478108
- J. Ostojić, C. Panozzo, J. Lasserre, C. Nouet, F. Courtin, C. Blancard, J. di Rago, and G. Dujardin, "The Energetic State of Mitochondria Modulates Complex III Biogenesis through the ATP-Dependent Activity of Bcs1", Cell Metabolism, vol. 18, pp. 567-577, 2013. http://dx.doi.org/10.1016/j.cmet.2013.08.017
- M. de Taffin de Tilques, D. Tribouillard-Tanvier, E. Tétaud, E. Testet, J. di Rago, and J. Lasserre, "Overexpression of mitochondrial oxodicarboxylate carrier (ODC1) preserves oxidative phosphorylation in a yeast model of the Barth syndrome", Disease Models & Mechanisms, 2017. http://dx.doi.org/10.1242/dmm.027540
- R.S. Aiyar, M. Bohnert, S. Duvezin-Caubet, C. Voisset, J. Gagneur, E.S. Fritsch, E. Couplan, K. von der Malsburg, C. Funaya, F. Soubigou, F. Courtin, S. Suresh, R. Kucharczyk, J. Evrard, C. Antony, R.P. St.Onge, M. Blondel, J. di Rago, M. van der Laan, and L.M. Steinmetz, "Mitochondrial protein sorting as a therapeutic target for ATP synthase disorders", Nature Communications, vol. 5, 2014. http://dx.doi.org/10.1038/ncomms6585
- M.G. Baile, M. Sathappa, Y. Lu, E. Pryce, K. Whited, J.M. McCaffery, X. Han, N.N. Alder, and S.M. Claypool, "Unremodeled and Remodeled Cardiolipin Are Functionally Indistinguishable in Yeast", Journal of Biological Chemistry, vol. 289, pp. 1768-1778, 2014. http://dx.doi.org/10.1074/jbc.M113.525733
- X. Wang, X. Zuo, B. Kucejova, and X.J. Chen, "Reduced cytosolic protein synthesis suppresses mitochondrial degeneration", Nature Cell Biology, vol. 10, pp. 1090-1097, 2008. http://dx.doi.org/10.1038/ncb1769
- A. Lebreton, C. Saveanu, L. Decourty, J. Rain, A. Jacquier, and M. Fromont-Racine, "A functional network involved in the recycling of nucleocytoplasmic pre-60S factors", The Journal of Cell Biology, vol. 173, pp. 349-360, 2006. http://dx.doi.org/10.1083/jcb.200510080
- S. Chen, Q. He, and M.L. Greenberg, "Loss of tafazzin in yeast leads to increased oxidative stress during respiratory growth", Molecular Microbiology, vol. 68, pp. 1061-1072, 2008. http://dx.doi.org/10.1111/j.1365-2958.2008.06216.x
- D. Acehan, Y. Xu, D.L. Stokes, and M. Schlame, "Comparison of lymphoblast mitochondria from normal subjects and patients with Barth syndrome using electron microscopic tomography", Laboratory Investigation, vol. 87, pp. 40-48, 2007. http://dx.doi.org/10.1038/labinvest.3700480
- X. Wang, and X.J. Chen, "A cytosolic network suppressing mitochondria-mediated proteostatic stress and cell death", Nature, vol. 524, pp. 481-484, 2015. http://dx.doi.org/10.1038/nature14859
- L. Wrobel, U. Topf, P. Bragoszewski, S. Wiese, M.E. Sztolsztener, S. Oeljeklaus, A. Varabyova, M. Lirski, P. Chroscicki, S. Mroczek, E. Januszewicz, A. Dziembowski, M. Koblowska, B. Warscheid, and A. Chacinska, "Mistargeted mitochondrial proteins activate a proteostatic response in the cytosol", Nature, vol. 524, pp. 485-488, 2015. http://dx.doi.org/10.1038/nature14951
- J. Sauerwald, T. Jores, M. Eisenberg-Bord, S.G. Chuartzman, M. Schuldiner, and D. Rapaport, "Genome-Wide Screens in Saccharomyces cerevisiae Highlight a Role for Cardiolipin in Biogenesis of Mitochondrial Outer Membrane Multispan Proteins", Molecular and Cellular Biology, vol. 35, pp. 3200-3211, 2015. http://dx.doi.org/10.1128/MCB.00107-15
- H. Kim, A.R. Grant, M.S. Simic, R.A. Kohnz, D.K. Nomura, J. Durieux, C.E. Riera, M. Sanchez, E. Kapernick, S. Wolff, and A. Dillin, "Lipid Biosynthesis Coordinates a Mitochondrial-to-Cytosolic Stress Response", Cell, vol. 166, pp. 1539-1552.e16, 2016. http://dx.doi.org/10.1016/j.cell.2016.08.027
- S.C. Johnson, M.E. Yanos, E. Kayser, A. Quintana, M. Sangesland, A. Castanza, L. Uhde, J. Hui, V.Z. Wall, A. Gagnidze, K. Oh, B.M. Wasko, F.J. Ramos, R.D. Palmiter, P.S. Rabinovitch, P.G. Morgan, M.M. Sedensky, and M. Kaeberlein, "mTOR Inhibition Alleviates Mitochondrial Disease in a Mouse Model of Leigh Syndrome", Science, vol. 342, pp. 1524-1528, 2013. http://dx.doi.org/10.1126/science.1244360
- X. Zheng, L. Boyer, M. Jin, Y. Kim, W. Fan, C. Bardy, T. Berggren, R.M. Evans, F.H. Gage, and T. Hunter, "Alleviation of neuronal energy deficiency by mTOR inhibition as a treatment for mitochondria-related neurodegeneration", eLife, vol. 5, 2016. http://dx.doi.org/10.7554/eLife.13378
- N. Segev, and J.E. Gerst, "Specialized ribosomes and specific ribosomal protein paralogs control translation of mitochondrial proteins", Journal of Cell Biology, vol. 217, pp. 117-126, 2017. http://dx.doi.org/10.1083/jcb.201706059
- M.S. Longtine, A. Mckenzie III, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle, "Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae", Yeast, vol. 14, pp. 953-961, 1998. http://dx.doi.org/10.1002/(SICI)1097-0061(199807)14:10<953::AID-YEA293>3.0.CO;2-U
- B. Guérin, P. Labbe, and M. Somlo, "[19] Preparation of yeast mitochondria (Saccharomyces cerevisiae) with good P/O and respiratory control ratios", Biomembranes Part F: Bioenergetics: Oxidative Phosphorylation, pp. 149-159, 1979. http://dx.doi.org/10.1016/0076-6879(79)55021-6
- O.H. LOWRY, N.J. ROSEBROUGH, A.L. FARR, and R.J. RANDALL, "Protein measurement with the Folin phenol reagent.", The Journal of biological chemistry, 1951. http://www.ncbi.nlm.nih.gov/pubmed/14907713
- M. Rigoulet, and B. Guerin, "Phosphate transport and ATP synthesis in yeast mitochondria", FEBS Letters, vol. 102, pp. 18-22, 1979. http://dx.doi.org/10.1016/0014-5793(79)80919-9
- R.K. Emaus, R. Grunwald, and J.J. Lemasters, "Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 850, pp. 436-448, 1986. http://dx.doi.org/10.1016/0005-2728(86)90112-x
- M. Rak, E. Tetaud, F. Godard, I. Sagot, B. Salin, S. Duvezin-Caubet, P.P. Slonimski, J. Rytka, and J. di Rago, "Yeast Cells Lacking the Mitochondrial Gene Encoding the ATP Synthase Subunit 6 Exhibit a Selective Loss of Complex IV and Unusual Mitochondrial Morphology", Journal of Biological Chemistry, vol. 282, pp. 10853-10864, 2007. http://dx.doi.org/10.1074/jbc.M608692200
- H. Schägger, and G. von Jagow, "Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form", Analytical Biochemistry, vol. 199, pp. 223-231, 1991. http://dx.doi.org/10.1016/0003-2697(91)90094-a
- G. Arselin, J. Vaillier, P. Graves, and J. Velours, "ATP Synthase of Yeast Mitochondria", Journal of Biological Chemistry, vol. 271, pp. 20284-20290, 1996. http://dx.doi.org/10.1074/jbc.271.34.20284
- M. Gassmann, B. Grenacher, B. Rohde, and J. Vogel, "Quantifying Western blots: Pitfalls of densitometry", ELECTROPHORESIS, vol. 30, pp. 1845-1855, 2009. http://dx.doi.org/10.1002/elps.200800720
- F. Vitello, and J. Zanetta, "Thin-layer chromatography of phospholipids", Journal of Chromatography A, vol. 166, pp. 637-640, 1978. http://dx.doi.org/10.1016/s0021-9673(00)95654-1
- F. Gonzalvez, Z.T. Schug, R.H. Houtkooper, E.D. MacKenzie, D.G. Brooks, R.J. Wanders, P.X. Petit, F.M. Vaz, and E. Gottlieb, "Cardiolipin provides an essential activating platform for caspase-8 on mitochondria", The Journal of Cell Biology, vol. 183, pp. 681-696, 2008. http://dx.doi.org/10.1083/jcb.200803129
AUTHOR CONTRIBUTIONS
D.T.-T., J.-P.L., M.d.T.d.T. F.G., E.S., R.K, M.B. and P.X. P. performed the experiments; D.T.-T., J.-P.L., J.-P.d.R., M.d.T.d.T. R. K, and E.T. analyzed the data; M.L.G. statistically analyzed the data of Fig. 2; D.T.-T., J.-P.L. and J.-P.d.R designed the research and wrote the paper.
ACKNOWLEDGMENTS
We gratefully thank the lipidomic platform of Bordeaux for lipid analysis on the yeast strains and J.J. Bessoule for helpful discussions, and to Emilia Kouroussis for English revisions.
This work was supported by grants from the Association Française contre les Myopathies (AFM) to J.-P.dR and D.T.-T., and P.X.P, the Barth Syndrome Founda-tion (BSF), Association Barth France to D.T.-T and the Associa-tion contre les Maladies Mitochondriales (AMMI) to D.T.-T. M.d.T.d.T. was supported by a PhD fellowship from the French Ministère de l’Enseignement et de la Recherche.
COPYRIGHT
© 2018

Decreasing cytosolic translation is beneficial to yeast and human Tafazzin-deficient cells by de Taffin de Tilques et al. is licensed under a Creative Commons Attribution 4.0 International License.








