
Reviews:
Microbial Cell, Vol. 2, No. 11, pp. 412 - 428; doi: 10.15698/mic2015.11.237
Electron microscopy for ultrastructural analysis and protein localization in Saccharomyces cerevisiae

Department of Cell Biology, University Medical Center Groningen, University of Groningen, Groningen, The Netherlands.
Keywords: electron microscopy, electron tomography, immunolabeling, chemical fixation, cryo-immobilization, correlative light and electron microscopy, Saccharomyces cerevisiae.
Abbreviations:
CLEM - correlative light-electron microscopy,
CEMOVIS - cryo-electron microscopy of vitreous sections,
EM - electron microscopy,
ER - endoplasmic reticulum,
ET - electron tomography,
FS - freeze substitution,
GA - glutaraldehyde,
HPF - high-pressure freezing,
IEM - immunoelectron microscopy,
LC - lead citrate,
SEM - scanning electron microscopy,
TA - tannic acid,
TEM - transmission electron microscopy,
UA - uranyl acetate.
Received originally: 24/06/2015 Received in revised form: 23/08/2015
Accepted: 31/08/2015
Published: 12/10/2015
Correspondence:
Fulvio Reggiori, f.m.reggiori@umcg.nl
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Andri Frankl, Muriel Mari and Fulvio Reggiori (2015). Electron microscopy for ultrastructural analysis and protein localization in Saccharomyces cerevisiae. Microbial Cell2(11): 412-428.
Abstract
The yeast Saccharomyces cerevisiae is a key model system for studying of a multitude of cellular processes because of its amenability to genetics, molecular biology and biochemical procedures. Ultrastructural examinations of this organism, though, are traditionally difficult because of the presence of a thick cell wall and the high density of cytoplasmic proteins. A series of recent methodological and technical developments, however, has revived interest in morphological analyses of yeast (e.g. [1][2][3]). Here we present a review of established and new methods, from sample preparation to imaging, for the ultrastructural analysis of S. cerevisiae. We include information for the use of different fixation methods, embedding procedures, approaches for contrast enhancement, and sample visualization techniques, with references to successful examples. The goal of this review is to guide researchers that want to investigate a particular process at the ultrastructural level in yeast by aiding in the selection of the most appropriate approach to visualize a specific structure or subcellular compartment.
INTRODUCTION
The yeast Saccharomyces cerevisiae is an invaluable model system for the investigation of many biological processes but also for certain ultrastructural aspects of the eukaryotic cells. It is perhaps one of the most widely employed model organisms for research in life sciences disciplines because of its amenability to genetic and biochemical approaches. By studying the yeast counterparts of mammalian proteins S. cerevisiae helped to determine the function of countless proteins important in human biology. As genomic projects continue to provide increasing amounts of high throughput datasets about the potential regulation and function of genes, the challenge is to assign a molecular role to the corresponding gene products and determine their overall contribution to the cell physiology. For this goal, researchers take advantage of a multitude of experimental approaches and methods. One of them is electron microscopy (EM), which allows the analysis of the ultrastructure of cells and tissues, and also of purified subcellular compartments. EM helps to study cellular processes such as cytoskeleton organization, transport vesicle formation and the establishment of organelle architecture. It also contributes to the precise localization of proteins and other cellular components. Ultrastructural EM methods rely on microscopes that use electrons to obtain images at a higher resolution than those generated by microscopes. This is due to the fact that the wavelengths of electrons are much shorter than those of the photons used by light microscopes, and consequently the resolving power is much better (up to 10 Angstrom versus approximately 200 nm).
–
In the past decade, innovations and breakthroughs turned EM from a mainly pure morphological approach to a much broader one, especially through integration of a variety of immunocytochemical and correlative light-electron microscopy techniques. In addition to their high resolution, another unique aspect of EM methods is that they provide information about the cellular context of the structure of interest, which very often cannot be explored with other experimental approaches. This advantage becomes even clearer when analyzing mutant cells, for the reason that EM can provide clues about the possible function of a protein and the effects caused by its mutation. The combination of EM and yeast genetics, which easily permits the knockout of a gene or the generation of point mutants, has great investigative potentials.
–
This potential, however, has only been minimally exploited mainly because yeast represents a challenge for most EM procedures. It possesses a cell wall, which impairs cell infiltration with chemicals and resins, and its high protein concentration in the cytoplasm, which makes it difficult to obtain good contrast and morphological resolution. Nevertheless, a series of recent EM developments and adaptations started a new era for ultrastructural investigations in this organism. Although, there is a myriad of different EM techniques (Figure 1), ranging from sample preparation to image analysis, it is often difficult to decide which could be the most appropriate approach to answer a specific biological question.
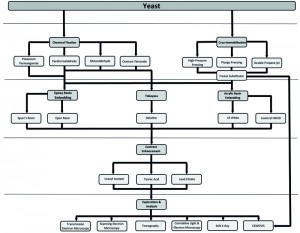 | FIGURE 1: EM approaches to explore yeast ultrastructure and immunocytochemical localization of proteins. Schematic representation summarizing mainstream approaches for EM and IEM (immunoelectron microscopy) to explore the morphology and protein localization in yeast. |
We describe and discuss techniques that have been successfully applied for yeast, and provide information to select the optimal EM method for specific research questions. While this compendium is focused on S. cerevisiae, most of the presented approaches are applicable to other unicellular yeast such as Schizosaccharomyces pombe, Candida albicans and Pichia pastoris, and they are also valid for the analysis of filamentous fungi.
METHODS FOR YEAST CELL FIXATION
The primary goal of sample fixation is to immobilize cellular structures in a way that they remain preserved as close as possible to their native state inside a living cell. To achieve this goal a number of chemical and physical fixation methods are available. Every fixation method comes with its own advantages and disadvantages, which have to be considered depending on the goal of the analysis (Table 1). Fixation is frequently followed by the removal of water, which requires that cellular structures are stabilized sufficiently to prevent their extraction along with the water. This is done through dehydration, usually performed by using organic solvents such as ethanol, methanol or acetone. The water removal from the sample is necessary to allow subsequent infiltration with a structural support, often a resin, which can be polymerized to provide rigidity to the specimen to endure the electron beam in the electron microscope. This last step is also essential to obtain solid blocks of cells that can be easily cut and stained for EM. This general approach is commonly considered the conventional EM procedure for sample preparation.
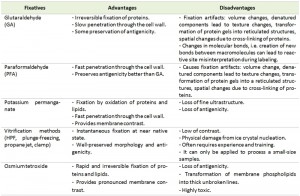 | TABLE 1. Advantages and disadvantages of different types of fixation. |
The most common form of fixation for yeast is the chemical cross-linking of proteins and lipids. When performing immunoelectron microscopy (IEM) this type of immobilization is generally limited to aldehydes because they minimally alter epitopes. Fixation is generally followed by either osmium tetroxide or potassium permanganate treatments when using conventional embedding procedures with epoxy resins (see below). Chemical fixation is not an instantaneous process and consequently specific organelles such as vacuoles, which are mostly composed of water, require time to be completely immobilized and thus their morphology is frequently altered from its native state.
–
Due to the chemical reactions taking place during fixation, a release of protons often changes the pH, which may impede optimal cross-linking and may also affect the subsequent embedding of the sample [4]. To overcome this problem, fixatives are often delivered in buffered solutions and the most frequently employed ones to maintain the neutrality are phosphate, cacodylate or PIPES buffer [5]. PIPES and cacodylate buffers enhance membrane preservation via addition of calcium ions [6]. While cacodylate buffers are based on arsenic, PIPES is not. It is suggested that organic buffers such as PIPES and PHEM improve cell preservation and limit the formation of electron dense precipitates compared to non-organic buffer including phosphate and cocadylate [7][8][9][10]. Each buffer, in combination or not with other cations, can give a better ultrastructural preservation of specific structures [4][11].
–
The yeast cell wall is a significant obstacle for optimal fixation. This structure, which surrounds the plasma membrane, is a rigid extracellular polymer composed of mannoproteins, glucans and chitin [12]. The reduced porosity and cross-reactions between the components of the cell wall and the added reagents make yeast cell infiltration with embedding mixtures slow and inefficient [10][13]. The enzymatic removal of the cell wall with glucanases such as glusulase, lyticase or zymolyase, prior to fixation reduces these problems but could affect the cell physiology [6][14]. An alternative is incubation with sodium metaperiodate, a step that can be introduced after fixation [13]. Metaperiodate breaks glycosylic bonds to release proteins from the cell wall, leaving it more permeable to viscous embedding solutions.
–
Aldehydes
Glutaraldehyde (GA) is one of the most common chemical fixatives used for EM and it is also employed extensively for yeast [6][10]. It irreversibly binds with amino groups, like those on lysine residues, and forms various intra- and inter-protein bridges. Although GA is able to react with other molecules such as specific carbohydrates and amide groups, this compound does not bind well to lipids and therefore it is often combined with another fixative that cross-links with lipids with higher efficiency [15]. One of the chemicals used for this aim is paraformaldehyde (PFA). Moreover the small size of PFA permits its rapid diffusion across the cell wall [16]. GA, as a five-carbon compound, is relatively large and uncharged in a solution, and therefore its diffusion through the yeast cell wall is slower [6].
–
In combination, PFA initiates stabilization of cellular structures until GA can begin to react. PFA targets similar amino groups as GA but as a fixative, it is relatively unstable and consequently its cross-linking is reversible. The end result of a combination of the two fixatives is a clear image with little or no extraction, although not always perfectly accurate due to some of the possible artifacts that come from chemical fixation such as breaks, kinks or blisters in membranes. Typically, both GA and PFA are used in concentration ranging from 0.05% to 5% [4][6], with PFA in higher concentrations than GA. The irreversibility and high cross-linking properties of GA lead to severe alterations of epitopes and consequently the concentration of this chemical must be kept to a minimum if immunocytochemical examinations are planned. Other aldehydes have also been used in yeast such as acrolein [17], which is often employed together with GA, as well as other chemicals including imidoesters and peroxydisulphates [11]. Currently these fixatives are rarely used especially because they do not preserve the morphology better than GA and/or PFA.
–
Potassium permanganate
Potassium permanganate (KMnO4) was one of the first fixatives to be used for EM [18]. Potassium permanganate is also a common post-fixative for yeast because unlike aldehydes, it better preserves lipid bilayers and it is thus employed alone or in combination with aldehydes [6]. Permanganate binding to lipids already provides some membrane staining. The overall membrane morphology with this type of fixation appears highly contrasted, however, closer observations show an extracted morphology. Although some prominent non-membranous structures, such as ribosome and microtubules, are not preserved and certain organelles like mitochondria and lipid droplets have a partially altered morphology, other subcellular compartments such as the endoplasmic reticulum (ER), nucleus, plasma membrane, vacuole, Golgi and endosomes are well defined in permanganate-fixed yeast preparations [6][19][20][21]. Potassium permanganate has been employed in a variety of concentrations in yeast, ranging from 0.5% to 6% [6] but this type of fixation is incompatible with immunolabeling due to heavily altered epitopes.
–
High-pressure freezing (HPF) and freeze substitution (FS)
The use of conventional chemical fixation can sometimes lead to artifacts. Therefore physical immobilization approaches have been implemented and most of them are based on rapid freezing of the sample. There are numerous ways to cryo-immobilize yeast and the major advantages of all these methods is that they are generally instantaneous and faster than conventional chemical fixation. These methods include plunge [22], impact [23], double-propane jet, self-pressurized freezing [24] and high-pressure freezing [25]. Their central principle is to vitrify (freeze the water without ice crystals formation) cells before further fixing them using chemicals. We will exclusively discuss HPF and FS because these are the most frequently used techniques, information on other types of quick freeze procedures can be found in other reviews [26][27].
–
HPF is currently the main approach for physical immobilization of yeast. Although other techniques such as plunge freezing and impact immobilization were more popular in the past, HPF is more reliable and efficient. Although it should be noted that it requires sophisticated and expensive equipment, this technique allows freezing relatively large quantities of yeast without cryo-protectants. HPF is achieved through application of high hydrostatic pressure and rapid lowering of the freezing point to halt the rate of ice crystal nucleation and growth [28][29]. This immobilizes the liquid milieu inside and outside the yeast in a vitreous near-native state. Although volumes of yeast up to approximately 120 mm3 can be high pressure frozen, a volume around 10-20 mm3 is considered a more reasonable working quantity [11]. Additional preservation of the native state can be achieved by adding non-penetrating cryo-protectants such as low melting point agarose or bovine serum albumin to the yeast suspension [30]. These two molecules have the ability to bind water through hydrogen bonds and thereby change its freezing properties, which further aids the freezing process [31]. Once the cells have undergone HPF, the water inside is extracted and substituted through a process known as freeze substitution (FS).
–
The principle of FS has been around for more than 40 years. The concept of dehydrating and fixing cells at very low temperatures for EM can be traced back to the 1960’s [32]. FS involves substitution of the water found inside cells, initially with an organic solvent, typically acetone, ethanol or methanol, and subsequently with a resin at low temperatures before finally increasing the sample temperature to room temperature [33][34]. Chemical fixatives are often added to the solvent employed for FS, to provide further immobilization during this procedure. Commonly used fixatives for FS mixtures include osmium tetroxide, uranyl acetate, PFA (0.1 – 3%) and GA (0.1 – 1%), sometime in combination with small amounts of water (0.1 – 5%) to increase membrane contrast [26].
–
Unlike conventional chemical fixation, the fixation steps of FS take place during or after the dehydration steps. Temperatures for FS vary between -90°C and -78°C, and the solvent will dissolve and replace the cell water over a period of hours. Fixatives are not very reactive at these low temperatures, but become homogenously distributed throughout the yeast cell despite the presence of a cell wall because of the long incubation periods (i.e. days), though rapid FS protocols have been developed as well [35]. The low temperatures keep the subcellular structure in place during the diffusion and the action of fixatives. At the moment the temperature allows the fixation to occur, the fixative is already in place, homogenously distributed.
–
Osmium tetroxide
This chemical is commonly used after an initial fixation with GA and/or PFA, and before embedding with a resin after FS. Osmium tetroxide binds lipids and promotes the oxidation of saturated bonds present in their fatty acid moieties causing retention of lipids in EM preparations [36]. Osmium tetroxide also adds density and contrast to lipid bilayers increasing the visibility of membranous compartments. This latter characteristic is due to the molecular structure of this compound, which possesses a high density allowing electron scattering.
–
Unlike permanganate and aldehydes, osmium tetroxide infiltrates the cell wall of yeast even less efficiently [14]. It is thus necessary to remove or permeabilise the cell wall for short incubations with osmium tetroxide to promote its penetration into the yeast cell [14]. Sample incubation times with osmium tetroxide have important effects on the preparation morphology. Too short exposures do not allow a good infiltration of the yeast and a sufficient fixation of lipids, resulting in lipid extraction that is visible as blank membrane profiles on micrographs [15]. Prolonged exposures, in contrast, cause the extraction of cell components especially during the subsequent dehydration steps as well as deposits of electron-dense osmium precipitates near membrane concentrations [15].
–
After an appropriate exposure time, between 15 and 60 minutes depending on which protocol is used, osmium tetroxide extracts cell components much less than potassium permanganate, leaving relatively small cellular components including microtubules, microfilaments, chromatin and ribosomes visible [6]. Osmium tetroxide is a commonly used fixative in yeast and particularly in combination with HPF followed by FS and sample embedding in Epon or Lowicryl HM20, it has been used to investigate for example the spindle pole body and nuclear envelope [37], the cell wall structure [38], and the formation of septa and nuclear pores [39].
–
Osmium tetroxide fixation is not recommended when cytochemical or immunocytochemical labeling are performed because the extractions, as well as the volume and morphological changes that it causes, can lead to physical distortions that greatly interfere with the preservation of enzyme activities and antibody-antigen reactions [11]. Nonetheless, in low quantities or with certain epitopes, osmium tetroxide can still be used without altering immunolabeling [22].
EMBEDDING APPROACHES: STRUCTURAL MORPHOLOGY VERSUS IMMUNOGOLD LABELING
Yeast, just like any other cell type, have to be properly infiltrated by a chemical compound that will generate the structural support required for both sectioning and viewing in an electron microscope. In choosing the resin, one must determine primarily what the focus of the study will be: structural morphology or protein localization, i.e. epitope preservation for immunolabeling. Moreover, the chosen resin has to be appropriate to the employed fixation (see below). There are two main categories of plastic resins: epoxy and acrylic resins. Epoxy resins, such as Epon or Spurr’s, are good for resolving the cell morphology whereas acrylic resins, such as LR White and Lowicryl HM20, are better in preserving antigenicity.
–
Epoxy resins
Epoxy resins initiated the age of fine structural analysis by EM; other methacrylate resins were only marginally successful [40][41]. The significant advantages of epoxy resins come from their ability to cross-link virtually all structures composing a cell without losing the plasticity required to produce ultrathin sections.
–
Epon mixtures were introduced for EM in yeast in the early 1960’s [40]. They continue to be used quite often as the primary embedding resin for yeast especially in combination with HPF and FS [6][25][26] (Figures 2A and 2C). It was employed to study septin rings during cell division [42] or the nucleus and vacuole connections [43]. Epon resins were used in combination with potassium permanganate- or GA-fixed yeast to study for example the ER morphology [44][45]. The components of a modern Epon mixture are the resin 812, the hardeners dodecenyl succinic anhydride and nadic methyl anhydride, and the accelerator benzyldimethylamine [15]. Application of heat (or ultraviolet light) to this solution yields a rigid 3-dimensional polymer that is both resistant to heat and solvents, and is structurally stable under electron beams.
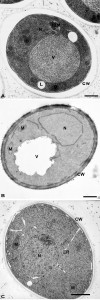 | FIGURE 2: Morphology of yeast cells embedded in epoxy resins. (A) Cells were cryofixed in liquid propane, freeze-substituted in acetone containing 4% OsO4 and embedded in Epon. CW, cell wall; N, Nucleus; IB, Inclusion body; IBM, Inclusion body with membrane; L, lipid droplets; V, Vacuole. Scale bar, 0.5 µm. This image was originally published in [46] © Springer. (B) Yeast was fixed with 1.5% KMnO4, dehydrated with acetone and embedded in Spurr’s resin. CW, cell wall; M, mitochondria; N, Nucleus; V, vacuole. Scale bar, 0.5 µm. This image was originally published in [47] © the American Society for Biochemistry and Molecular Biology. (C) Cells were high-pressure frozen, freeze-substituted in acetone, and embedded in a mixture of Epon-Spurr’s resin. CW, cell wall; ER, endoplasmic reticulum; M, mitochondria; N, nucleus. Scale Bar, 1.0 µm. This image was originally published in [48] © Elsevier Limited. |
Another epoxy resin is the Spurr’s (or vinylcyclohexene dioxide) resin. The di-epoxide groups present in this mixture produce high cross-linking [15]. Its low viscosity facilitates its infiltration into a variety of tissues that are difficult to embed including plant and yeast [46]. In particular, the cell wall of yeast can be efficiently infiltrated by the Spurr’s mixture and this allows an excellent preservation of the morphology, especially that of membranous structures, such as the ER, the nuclear envelop and the plasma membrane [6]. Excellent results have been obtained in combination with potassium permanganate (Figure 2B) to investigate the compartments of the secretory pathway, but also the morphology of mitochondria [47][48], vacuoles and autophagosomes [49], and the endocytic intermediates [50]. Spurr’s resin has also been used to study compartments of the secretory and endosomal systems in cells fixed with either GA (followed by enzymatic removal of the cell wall) or plunge freezing [51][52][53][54][55] (Figure 2C).
–
As mentioned above, both Epon and Spurr’s resins allow excellent resolution of yeast morphology in combination with the appropriate fixation methods. Epoxy resins, however, present a major disadvantage because they are typically not compatible with immunocytochemical reactions aimed at localizing proteins. Due to their extremely hydrophobic nature, samples have to be completely dehydrated at room temperature or above, using solvents that can often cause protein denaturation. Furthermore the high polymerization temperatures (above 50°C) lead to further epitope denaturation. This limitation can be circumvented when studying the endo-lysosomal system through pre-embedding labeling [56] but this approach has not been applied to yeast.
–
Acrylic resins
Limitations of the epoxy resins for immunocytochemical studies prompted a dedicated attempt to create more hydrophilic resins that combine good cutting properties and electron-beam resistance. A major breakthrough came with the introduction of Lowicryl mixtures like e.g. Lowicryl HM20, which are partly hydrophilic and consequently efficiently penetrate tissues [11]. A mixture of aliphatic acrylates and methacrylate esters composes Lowicryl resins, which have a low viscosity and therefore they efficiently infiltrate the cell wall of yeast [11]. Lowicryl HM20 was developed in 1986 [57] initially to be able to handle much lower polymerization temperatures required (below -50°C) for HPF-FS approaches. Importantly, it appears that any remaining water acts as a support agent at low temperatures to stabilize protein conformation during the dehydration process [58]. The fact that lower temperatures significantly decrease the negative effects of dehydration on structural preservation and epitope denaturation, as well as negligible lipid extraction, makes immunolabeling reactions on Lowicryl resin-embedded samples more effective [59][60]. In yeast, Lowicryl HM20 has been employed in combination with either chemical (Figure 3A) or HPF fixation to immunolocalize for example vacuolar enzymes [13] or proteins accumulated in the ER [61].
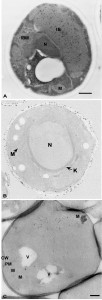 | FIGURE 3: Morphology and immunolabeling of yeast cells embedded in acrylic resins or processed following the Tokuyasu method. (A) Yeast was cryofixed in propane, freeze-substituted in acetone containing 3% GA and embedded in Lowicryl HM20 at low temperatures. Specific antibodies and protein A were used to localize the COX complex. IB, inclusion body; IBM, inclusion body with membrane; M, mitochondria; N, nucleus. Scale bar, 0.5 µm. This image was originally published in [46] © Springer. (B) Cells were fixed in GA/PFA, dehydrated with ethanol and embedded in LR White resin. Immunolabeling was directed to cell wall antigens. K, karmellae; M, mitochondria; N, nucleus. This image was originally published in [6] © John Wiley and Sons. (C) Cells were fixed with 4% PFA and 0.4% GA, treated with sodium metaperiodate, embedded in 12% gelatin and infiltrated with 2.3 M sucrose before being frozen in liquid nitrogen. Atg9 was localized with antibodies and protein A-gold. CW, cell wall; M, mitochondria; PM, plasma membrane; V, vacuole. Scale bar, 0.5 µm. This image was originally published in [65] © Mari et al, 2010. |
Another polyhydroxy-aromatic acrylic resin is LR White [62]. This low viscosity mixture requires tissue dehydration before infiltration and allows a rapid embedding. Its polymerization can be initiated in different ways, i.e. by either heating to temperatures above 50°C, exposure to UV or addition of an aromatic tertiary amine that accelerates chemical reactions. LR White resin has also been used for a number of studies using chemically (Figure 3B) or physically fixed yeast to localize through immunological reactions nuclear pore complex subunits [63], plasma membrane Gas1 [64], endosomal proteins [65][66], endocytosed factors [67], actin [68], ER [69] and spindle body components [70].
–
The Tokuyasu method
The thawed-frozen section technique is better known as the Tokuyasu method, from its developer’s name [71][72]. This approach utilizes ultra-thin sections that are obtained by cryo-ultramicrotomy from material that has been chemically fixed by aldehydes, embedded in gelatin and frozen in liquid nitrogen. Immunolabeling and imaging of the sections, however, are done at room temperature. This technique has many advantages over other embedding procedure because it provides a high-resolution of membranes as well as a higher efficiency of immunological reactions [73]. The Tokuyasu method remains one of the most sensitive post-sectioning techniques for immunolabeling because aldehyde fixation is the only denaturing step for antigens (i.e. samples are not treated with organic solvents).
–
This method has recently been optimized for yeast [74] (Figure 3C). The major modification in the protocol has been the introduction of a post-fixation treatment with metaperiodate to promote an infiltration of gelatin. Since cryo-sections obtained with the Tokuyasu method are not contrasted using a negative staining but rather with uranyl acetate and lead citrate (see below), the extraction of the non-optimally fixed lipids and the high protein concentration in the cytoplasm create negative contrast that leads to a unique resolution of the yeast morphology [74].
–
The Tokuyasu method adapted to yeast has been successfully used to perform localization studies on mitochondria [75][76], endosomes [77][78], subdomains of the plasma membrane [79], nuclear pores [80] and autophagosomal membranes [81][82][83]. Lipids tend to be extracted during the preparation of cryo-sections because they are not optimally fixed and therefore structures like lipid droplets with membranes low in protein concentrations are not optimally preserved. A way to overcome this problem and other possible fixation artifacts is that the Tokuyasu technique is not restricted to chemical fixation but it can also be combined with physical immobilization by HPF followed by FS and a rehydration step [84]. This approach appears to work with yeast samples as well [74].
MEMBRANE CONTRASTING METHODS
The gun of an electron microscope emits a beam of electrons with a particular wavelength that depends on the acceleration voltage applied. A phosphor-coated screen makes the electrons passing through sections visible by absorbing them. This results in an image being drawn by the density of the sample staining and the resulting intensity (number) of electrons hitting the phosphor-coated screen. The components and structures present in biological samples have generally very little differences in density and consequently the contrasts in the image formed are minimal. Therefore, it is important to increase contrast in the sample (Table 2). This can be achieved by increasing the densities of structures by binding heavy metal salts to them. There are two main approaches to stain EM samples with heavy metals: (i) positive staining exhibits a positive contrast by increasing the density of a particular biological structure rather than any contiguous surrounding area; (ii) negative staining through heavy metal salts increases the density of the area around a specific molecular structure so that the structure of interest appears lighter than the surrounding material. It must be noted that there are also contrasting methods such as tannic acid (TA) staining that do not rely on heavy metal salts [85]. Generally staining is carried out once sections have been cut and any immunocytochemical labelling has been performed.
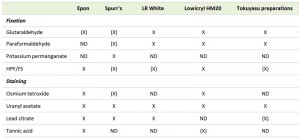 | TABLE 2. Combinations of embedding media, fixation methods and staining procedures employed for yeast ultrastructural analyses. X, frequently used combination; (X), Combination not often used; ND, never done. |
Positive staining
The two most common compounds used for the positive contrasting are uranyl acetate (UA) and lead citrate (LC). The staining mechanisms of these chemicals are not completely understood. Uranyl ions may be strongly attracted to phosphate and specific amino groups, which facilitates the identification of nucleic acids and proteins [86]. In contrast it is thought that lead ions bind to mostly negatively charged molecules such as hydroxyl groups or areas that have reacted to osmium tetroxide [87]. UA and LC are thus considered non-specific as they stain numerous different cellular components [15] and because of their complementary reactivity, they are often employed in combination to obtain better contrast. UA and LC are compatible with all the types of fixation and sample embedding, and the vast majority of EM analyses of yeast but also other organisms use these two heavy metal salts to contrast membranes.
–
Negative staining
Negative stains are often made from heavy metal salts such as uranyl, tungsten or molybdenum [15]. The heavy metal staining does not affect the macromolecular structures themselves, as with positive staining procedures, but rather the surrounding area. This results in a specimen that appears to be in negative contrast, i.e. a lighter tone against a darker background [15]. Although commonly used to identify small structures such as viruses, bacteria or little organelles, it can also be employed for the analysis of organisms of small size such as yeast [88]. The major advantage of negative staining compared to positive staining is that it highlights the structure of interest, especially when of small dimensions, without staining the structure itself, something that could alter its fine ultrastructural details. This aspect has been exploited in yeast to study glucan polymer formation during the regeneration of the cell wall in protoplasts [21][89] and protein filaments [90].
–
Tannic acid staining
Since it was first utilized as a mordant, i.e. a chemical that both fixes a dye on a cellular component and forms an insoluble compound with the dye, TA has become widely spread in its use because it optimally fixes a variety of different tissues and cells either by itself or in conjunction with GA [91]. TA in particular binds with high affinity to collagen, glycogen and various other subcellular complexes. Although it acts as a fixative, its mordant properties are very useful to greatly enhance the sample contrast [85]. As one of the few alternatives to the use of heavy metal salts, TA appears to avoid regions that would be stained by conventional contrasting agents such as UA. Therefore its use, alongside other staining agents, provides different contrasting patterns depending on the combination. In yeast, TA has only marginally been employed mostly as a post-embedding contrasting agent, to analyze purified microtubules [92] and nuclei [93] and COPII-coated vesicles [94].
2D AND 3D VISUALIZATION TECHNIQUES
Once the yeast samples are prepared, a variety of different imaging methods that revolve around the basic principles of EM are available, from widespread and fundamental approaches like transmission electron microscopy (TEM) and scanning electron microscopy (SEM), to more sophisticated techniques like correlative light-electron microscopy (CLEM), electron tomography (ET), cryo-electron microscopy of vitreous sections (CEMOVIS) and soft X-ray tomography. The analysis method largely determines the procedure of sample preparation and the type of data that are extracted from the sample. Each technique has its own strength and weakness and it is wise to carefully consider the research goal before opting for a particular approach.
–
Transmission electron microscopy
TEM is the most commonly employed form of EM, it has a resolution hundreds of times higher than that of the classical light microscopes and consequently it can visualize macromolecular structures and organelles that compose the cell at the nanoscale level. It consists of an interconnected set of electromagnetic lenses that channel a beam of primary electrons towards the sample [15]. As the primary electrons pass through the sample, they create a two-dimensional (2D) projection image with fine structural details [6]. TEM is easily handled by relatively inexperienced operators and can give some of the most detailed and high quality images that can be obtained [95]. With its high magnification and resolution, TEM makes it possible to see many of the structures present inside a yeast cell [95], which are not detectable and/or identifiable through light microscopy approaches including super-resolution ones. As TEM is one of the most widely used forms of EM for biological samples, a vast variety of publications are available. A large number of them are about the morphological and functional characterization of subcellular compartments of yeast processed for EM using preparation obtained with practically all the procedures presented in this review.
–
Electron tomography
ET is a method that generates three-dimensional (3D) reconstructions of a cellular structure, which provide more thorough and complete insights into its organization and possible functions. As a conventional electron micrograph has a large depth of focus and generates 2D projections, features in the z-axis of the section are superimposed on top of each other, making it hard to analyze and interpret them especially in thicker sections [96]. The improved insights are generated from a z-axis resolution that is at least 10 times better than the one of the average 2D projection image.
–
Initially an approach called serial sectioning was developed to introduce the third dimension in TEM, which has also been employed for studies in S. cerevisiae fixed with permanganate, GA/PFA or HPF before being infiltrated with an epoxy resin [97][98][99][100][101][102]. This technique involves the collection of several successive serial sections of the same sample, and then superposes and aligns their 2D images to generate 3D models. Some disadvantages for serial sectioning include a loss of material when handling cryo-sections and the stitching of multiple 2D projections can be very difficult. ET overcomes most of these issues and has the additional advantage of a simpler image collection and model reconstruction routine as well as a higher z-axis resolution. This technique combines a higher electron output with a tilt series of images created by rotating the specimen holder incrementally around a fixed axis [103]. The obtained tilt series of images (i.e. a collection of a large number of 2D images) are then stacked together and converted into a 3D representation of the sample.
–
ET has many of the same strengths and weaknesses as conventional TEM, but it is able to create a 3D image with a 1 – 10 nm resolution, which is similar to that of TEM (0.1 – 1 nm) and SEM (1 – 10 nm) [95]. It must be noted, however, that the 3D reconstructions created from ET tilt series of images are not complete representations. This is due to the limitation of the microscope sample holder that makes only possible to tilt the sample to a maximum of 60-70 degrees. This leaves the reconstructions with undefined cone shaped areas and consequently ET does not provide a complete 360 degrees overview of the zone of interest.
–
Alongside conventional TEM, ET has become a very prominent approach in many areas of cell biology and has been inclemently introduced in investigations performed in yeast as well. The most frequent approach has been to fix yeast through HPF and embedding in Epon, Spurr’s or Lowicryl HM20 resins before performing ET on thick sections that can range from 0.2 to 1 µm [21][26][104]. This type of methodology has, in between others, allowed studying the mitotic spindle/nuclear envelop [105][106], the septin rings formed between two dividing cells [42][107], multivesicular body formation [108][109][110], various aspects of mitochondrial ultrastructure [111][112][113], plasma membrane reshaping during endocytosis [114], and ER morphology [111][113]. Chemical fixation with permanganate followed by embedding in Epon resin has also been successfully used for electron tomography studies of lipid droplets [115]. Recently, 200 – 250 nm serial cryo-sections obtained with the Tokuyasu methods were resolved by ET and through immunolabeling proteins were localized in 3D reconstructions [1][75].
–
Scanning electron microscopy
SEM was developed approximately at the same time as the TEM. SEM can directly collect 3D representations of a cell surface or even an entire specimen but with lower resolution than TEM. SEM uses a focused de-magnified spot of electrons to scan over an electrically conductive specimen. The result of the electrons hitting the specimen is the release of a number of signals such as secondary electrons, backscatter electrons and X-rays [15]. Sensitive detectors that are specifically created for detecting them collect these various signals.
–
SEM is limited in terms of resolution at a high magnification when compared to conventional TEM (1-10 nm versus 0.1-1 nm, [95]) and thus it is usually employed to acquire information about the topology and morphology of the sample surface, rather than the internal morphology of a cell obtained by TEM. As a result, SEM is generally not used for EM-based immunolocalization studies but this is slowly changing with the introduction of new protocols and equipment [116]. However, there are a number of techniques that can be coupled to SEM to provide additional information. An example of these methodologies that is applied for the analysis of yeast is the focused ion beam-scanning electron microscopy (FIB-SEM), which allows the construction of 3D representations (with lower resolution than those ET), but permits the employment of much larger samples, up to 1 µm [117][118]. FIB-SEM has been exploited to generate 3D reconstructions of whole yeast cells, fixed with either GA/PFA and permanganate or HPF, before being embedded with a resin [119][120][121][122].
–
It should be noted that most biological specimens must be thoroughly dehydrated (i.e. critical point drying) and covered with a conductive metallic support film before being imaged by SEM. These treatments can distort cellular features and cause artifacts [15], though this does not apply to most embedded and sectioned samples due to the lower mass of specimen. There are several studies that relied on SEM of fixed and subsequently dehydrated yeast cells to examine surface features, i.e. cell wall and plasma membrane [123][124][125][126], but also cellular components like the nuclear pore complex is studied on isolated nuclei [93].
–
Correlative light-electron microscopy
The term CLEM includes all those methods that exploit light microscopy to localize structures of interest and subsequently determine ultrastructural details by EM (reviewed in [127][128][129][130]). These methods provide further insight into specific protein localizations that cannot be obtained by standard IEM because the immunological reaction does not allow the detection of the protein of interest [127]. Another application is the ultrastructural identification of a particular fluorescently labeled structure being monitored by fluorescent imaging. These latter approaches often require the fusion of the studied protein with a tag such as the green fluorescence protein (GFP), which can be visualized by fluorescent microscopy [131]. Subsequently, GFP is directly detected on the EM preparations if the employed fixation method and embedding support do not alter its ability to emit fluorescence upon excitation.
–
While some proteins have been optimized to retain their fluorescence capacity after EM-preparation [132], successful approaches employed fixation by either HPF followed by CEMOVIS (see below, [133][134][135]) or FS embedding in Lowicryl HM20 [2][114], or plunge-freezing before application of the Tokuyasu method [136]. Alternatively, GFP can be indirectly localized on EM preparations through either immunolabeling or chemical reactions if the tag consists of GFP fused with an enzyme that generates an electron dense precipitate, such as in the FLIPPER tag [3]. CLEM techniques have become very popular during the last decade and some have been applied to yeast studies. An example is the localization and characterization at the ultrastructural level of Sup35 prions with very good morphological results [137]. For this analysis yeast has been fixed with GA before being first imaged by fluorescence microscopy, and then processed for EM after OsO4 post-fixation and embedding into Epon812. Another example has been the analysis of actin filaments using the marker protein GFP-Abp1 [131].
–
Importantly, HPF and subsequent yeast cell embedding with Lowicryl HM20 allows to preserve the fluorescence of GFP in section and together with a new tool to correlate the fluorescence signal to EM preparations, this approach has successfully been used to study early endocytic events [2]. Tomographic CLEM analyses can also be performed on thick cryo-sections obtained using the Tokuyasu method and labeled then with antibodies conjugated to a fluorescent group [75]. Finally, CEMOVIS and soft X-ray tomography have also been employed for CLEM in yeast (see below).
–
Cryo-electron microscopy of vitreous sections
CEMOVIS (or cryo-EM/tomography) is a technique that employs vitrified biological samples and allows the observation of the specimen in a near native state [138][139][140]. Samples can range from cryo-sections of different thickness to entire cells if their width does not exceed 0.5 – 1 µm, and can be analyzed by TEM, SEM or ET. Moreover the serial sectioning of vitreous samples permits obtaining 3D reconstructions of larger samples [141]. The fact that samples are at a near native state infers that those chemicals, which could alter some ultrastructural details, have not been used [95].
–
CEMOVIS has, however, some downsides. Vitrified samples are unstained and therefore the contrast gained during the preparations of the sections is extremely low [95]. Because of this limitation and the fact that CEMOVIS requires sophisticated equipment (HPF system, cryo-ultramicrotome, cryo-EM, cryo-holder for ET…) as well as a high degree of technical expertise, published works exploiting this approach are scarce but are steadily increasing especially due to the major availability in HPF technologies, which overcome the previous use of plunge or slam freezing for sample vitrification. So far yeast has been exclusively used for proof-of-principle demonstrations for methods to be applied with CEMOVIS [133][134][135], but the resolution degree shown is very promising. Another disadvantage of CEMOVIS is that frozen preparations cannot be immunolabeled, which limits localization studies. Fluorescently tagged fusion proteins, however, are optimally preserved and as a result fluorescence signals can potentially be used for CLEM investigations [142].
–
Soft X-ray tomography
This method combines the features of light and electron microscopy. It is an easy and high throughput technique (similarly to light microscopy) that allows collecting low-resolution, absorption-based images similarly to EM [143]. Soft X-ray tomography permits a user to view a whole hydrated cell, and to examine its morphology at a high spatial resolution (0.8 µm) up to 15 µm deep. It is based on the principle of X-rays being absorbed directly by the different cellular components and the resulting image is practically a projection of the dose of X-rays passing through the specimen [144][145][146]. Organelles inside the cell are visualized directly due to their different biochemical composition and density. For example, a compartment with a high lipid content is much more sensitive to X-rays than an organelle that contain a significant portion of water such as the vacuole [147].
–
One of the big advantages of the soft X-ray tomography is its circumvention of the use of potentially damaging fixation and staining procedures because samples are cryo-immobilized. Another positive consequence of this feature is that this technique also allows performing CLEM examinations through the determination of the subcellular distribution of molecules tagged with a fluorescent label in entire and intact yeast cells projected in 3D [148]. On the downside of this approach is the relatively low resolution of the images, 50 nm at the maximum, which can vary depending on the machine used and the analyzed structure [149]. The 3D reconstructions, however, are accurate. While soft X-ray tomography requires special software and a very sensitive machinery able to detect the X-rays passing through the yeast samples, this technique is very helpful to determine organelle position, quantity, and structural changes due to growth conditions or mutations in large cell populations [147][143][150][151]. For example this approach has been used to demonstrate that the volumetric ratios between organelles such as the nucleus, nucleoli, mitochondria, vacuoles and lipid particles do not change throughout the cell cycle [150] or identifying factors required to regulate the shape of the mitotic nucleus [152].
CONCLUSIONS
EM has considerably contributed to the field of biological sciences over the past 60 years. Virtually every organelle and major structure of the cell has been discovered and characterized by EM. This has allowed researchers unraveling morphological details of healthy cells and the changes that they undergo in diseased or mutated states.
–
To determine which EM approach to use, it is crucial to consider the end goal of the research question. Choosing a particular imaging method, i.e. TEM, SEM or tomography will already reduce the available options for a researcher. As shown in Figure 1 and Table 2, there are many possibilities and no single defined route for sample preparation and analysis is universally applicable to solve all the questions. Different sample preparation steps can be combined or interchanged. For example one can employ HPF for cryoimmobilisation before either rehydrating the cells and using the Tokuyasu method or embedding them in a resin such as Epon. Therefore it is important to determine whether topographical details or conventional TEM images are required, or whether immunocytochemical methods will be employed to localize proteins. Another relevant aspect to consider is which subcellular organelle or structure will be examined.
–
Differences in lipid and protein composition, concentration and density can lead the cell components to be differently preserved and resolved depending on the employed fixation method, embedding support and contrasting agent. Sometimes it is better to choose a sample preparation method that is not ideal for immunological reactions but that provides a better morphology of the labeled structures rather than having a sample ideal for protein localization where the structure of interest is not clearly defined.
–
One of the advantages of EM approaches is that there are several alternatives to choose from. A major obstacle in combining different EM methods, however, will be the eventual availability and accessibility to a specific EM instrument.
–
We also wish to emphasize that ultrastructural observations must contain statistical evaluation. Rigorous stereological methods, including unbiased sampling techniques, can provide very precise quantifications about the subcellular distribution of a protein or compartment, the surface or volume of an organelle, or the recurrence of a phenotype [153][154][155]. To this aim, few guidelines have to be kept in consideration when designing and realizing EM analyses. Experiments have to be performed in triplicate and countings have to be done randomly. Typically 50-100 cells per experiment have to be examined but this number has to be increased if what is counted is infrequent. When carrying out IEM, two critical controls evaluating the specificity of the used antibodies have to be included to get an accurate estimation of the relative distribution of a protein. The first is to immunolabel sections prepared from cells not expressing the analyzed protein. The second is to perform an immunolabeling reaction that does not include the primary antibody.
–
As important as the invention of the electron microscope was, its continuous development and the integration of other specialized techniques and hardware are what makes cellular morphology a real corner stone of modern research. As a result of ongoing developments and improvements of computer software, electron detection systems, image enhancement solutions, automated quantification, new CLEM probes, multifunctional EM machines and data storage, EM has a bright future and it will move from a relative small-scale to large throughput type of analysis. Moreover machines and procedures that have recently been developed and used in other cell types such as serial section SEM [156] and serial block face SEM [157] or CLEM tags like miniSOG [156] or APEX [158], could also be applied to yeast. These advances will also result in more objective- and quantitative- studies than ever. Ultrastructural research in yeast will also benefit from these progresses and the continuous adaption and improvement of the established and new EM protocols for other systems will make ultrastructural studies in this model system a routine approach for investigators. In addition to having a large collection of mutant and knockout strains, yeast strains provide the straightforward possibility of endogenous gene fusion with sequences encoding for protein tags [156]. As a result, the development of new primary antibodies for IEM analyses is not an absolute necessity because it is sufficient to purchase a commercially available secondary antibody recognizing the tag and known to work for ultrastructural studies.
References
-
M. Mari, W.J. Geerts, and F. Reggiori, "Immuno‐ and Correlative Light Microscopy‐Electron Tomography Methods for
3D Protein Localization in Yeast", Traffic, vol. 15, pp. 1164-1178, 2014. http://dx.doi.org/10.1111/tra.12192 - W. Kukulski, M. Schorb, S. Welsch, A. Picco, M. Kaksonen, and J.A. Briggs, "Correlated fluorescence and 3D electron microscopy with high sensitivity and spatial precision", Journal of Cell Biology, vol. 192, pp. 111-119, 2011. http://dx.doi.org/10.1083/jcb.201009037
- J. Kuipers, T.J. van Ham, R.D. Kalicharan, A. Veenstra-Algra, K.A. Sjollema, F. Dijk, U. Schnell, and B.N.G. Giepmans, "FLIPPER, a combinatorial probe for correlated live imaging and electron microscopy, allows identification and quantitative analysis of various cells and organelles", Cell and Tissue Research, vol. 360, pp. 61-70, 2015. http://dx.doi.org/10.1007/s00441-015-2142-7
- C.H. Fox, F.B. Johnson, J. Whiting, and P.P. Roller, "Formaldehyde fixation.", Journal of Histochemistry & Cytochemistry, vol. 33, pp. 845-853, 1985. http://dx.doi.org/10.1177/33.8.3894502
- M. Schliwa, and J. van Blerkom, "Structural interaction of cytoskeletal components", The Journal of Cell Biology, vol. 90, pp. 222-235, 1981. http://dx.doi.org/10.1083/jcb.90.1.222
- R. Wright, "Transmission electron microscopy of yeast.", Microscopy research and technique, 2000. http://www.ncbi.nlm.nih.gov/pubmed/11169854
- P.S. Baur, and T.R. Stacey, "The use of PIPES buffer in the fixation of mammalian and marine tissues for electron microscopy", Journal of Microscopy, vol. 109, pp. 315-327, 1977. http://dx.doi.org/10.1111/j.1365-2818.1977.tb01145.x
- R.I. Schiff, and J.F. Gennaro, "The role of the buffer in the fixation of biological specimens for transmission and scanning electron microscopy", Scanning, vol. 2, pp. 135-148, 1979. http://dx.doi.org/10.1002/sca.4950020304
- R. Schultz, and E. Whitter, "Improved Procedures for Pineal Gland Fixation for Electron Microscopy", Journal of Pineal Research, vol. 6, pp. 267-284, 1989. http://dx.doi.org/10.1111/j.1600-079X.1989.tb00423.x
- J. Mulholland, and D. Botstein, "Immunology microscopy of aldehyde-fixed yeast cells", Guide to Yeast Genetics and Molecular and Cell Biology Part C, pp. 50-81, 2002. http://dx.doi.org/10.1016/S0076-6879(02)51841-3
-
J.N. Skepper, "Fine Structure Immuno‐cytochemistry. By G
areth Griffiths ", Journal of Microscopy, vol. 172, pp. 93-93, 1993. http://dx.doi.org/10.1111/j.1365-2818.1993.tb03398.x - G. Lesage, and H. Bussey, "Cell wall assembly in Saccharomyces cerevisiae.", Microbiology and molecular biology reviews : MMBR, 2006. http://www.ncbi.nlm.nih.gov/pubmed/16760306
- E. van Tuinen, and H. Riezman, "Immunolocalization of glyceraldehyde- 3-phosphate dehydrogenase, hexokinase and carboxypeptidase Y in yeast cells at the ultrastructural level", Ultramicroscopy, vol. 21, pp. 202, 1987. http://dx.doi.org/10.1016/0304-3991(87)90132-X
- B. Byers, "Cytology of the yeast life cycle", Cold Spring Harbor Monograph Archive 1159-96., 1981.
- J. Bozzola, and L. Russell, "Electron Microscopy", 2nd Edition ed. Massachusetts: Jones & Bartlett Publishers., 1992.
- J.H. Bowes, and C.W. Cater, "The reaction of glutaraldehyde with proteins and other biological materials*", Journal of the Royal Microscopical Society, vol. 85, pp. 193-200, 1966. http://dx.doi.org/10.1111/j.1365-2818.1966.tb02180.x
- D.W. Schwab, A.H. Janney, J. Scala, and L.M. Lewin, "Preservation of fine Structures in Yeast by Fixation in a Dimethyl Sulfoxide-Acrolein-Glutaraldehyde Solution", Stain Technology, vol. 45, pp. 143-147, 1970. http://dx.doi.org/10.3109/10520297009067469
- J.H. Luft, "Permanganate—A New Fixative for Electron Microscopy", The Journal of Cell Biology, vol. 2, pp. 799-802, 1956. http://dx.doi.org/10.1083/jcb.2.6.799
- N. MACLEAN, "ELECTRON MICROSCOPY OF A FISSION YEAST, SCHIZOSACCHAROMYCES POMBE.", Journal of bacteriology, 1964. http://www.ncbi.nlm.nih.gov/pubmed/14234806
- N.J.W. Kreger-van Rij, and M. Veenhuis, "Bipolar budding in yeasts — an electron microscope study", Antonie van Leeuwenhoek, vol. 37, pp. 125-136, 1971. http://dx.doi.org/10.1007/BF02218473
- M. Osumi, "Visualization of yeast cells by electron microscopy", Microscopy, vol. 61, pp. 343-365, 2012. http://dx.doi.org/10.1093/jmicro/dfs082
- R. Ding, K.L. McDonald, and J.R. McIntosh, "Three-dimensional reconstruction and analysis of mitotic spindles from the yeast, Schizosaccharomyces pombe.", The Journal of cell biology, vol. 120, pp. 141-151, 1993. http://dx.doi.org/10.1083/jcb.120.1.141
- M. Hirsch, P. Montcourrier, Y. Pouliquen, and J. Escaig, "Quick-freezing technique using a ‘slamming’ device for the study of corneal stromal morphology", Experimental Eye Research, vol. 34, pp. 841-845, 1982. http://dx.doi.org/10.1016/S0014-4835(82)80045-6
- H. Han, J. Huebinger, and M. Grabenbauer, "Self-pressurized rapid freezing (SPRF) as a simple fixation method for cryo-electron microscopy of vitreous sections", Journal of Structural Biology, vol. 178, pp. 84-87, 2012. http://dx.doi.org/10.1016/j.jsb.2012.04.001
- K. McDonald, and T. Müller-Reichert, "Cryomethods for thin section electron microscopy", Guide to Yeast Genetics and Molecular and Cell Biology Part C, pp. 96-123, 2002. http://dx.doi.org/10.1016/S0076-6879(02)51843-7
- K. McDonald, "Cryopreparation Methods for Electron Microscopy of Selected Model Systems", Methods in Cell Biology, pp. 23-56, 2007. http://dx.doi.org/10.1016/s0091-679x(06)79002-1
- S. Murray, "Chapter 1 High Pressure Freezing and Freeze Substitution of Schizosaccharomyces pombe and Saccharomyces cerevisiae for TEM", Methods in Cell Biology, pp. 3-17, 2008. http://dx.doi.org/10.1016/S0091-679X(08)00401-9
- H. Moor, "Theory and Practice of High Pressure Freezing", Cryotechniques in Biological Electron Microscopy, pp. 175-191, 1987. http://dx.doi.org/10.1007/978-3-642-72815-0_8
- H. Moor, and K. Mühlethaler, "FINE STRUCTURE IN FROZEN-ETCHED YEAST CELLS", The Journal of Cell Biology, vol. 17, pp. 609-628, 1963. http://dx.doi.org/10.1083/jcb.17.3.609
- J. DUBOCHET, "High-pressure freezing for cryoelectron microscopy", Trends in Cell Biology, vol. 5, pp. 366-368, 1995. http://dx.doi.org/10.1016/s0962-8924(00)89071-6
- K. McDonald, H. Schwarz, T. Müller-Reichert, R. Webb, C. Buser, and M. Morphew, "“Tips and Tricks” for High-Pressure Freezing of Model Systems", Methods in Cell Biology, pp. 671-693, 2010. http://dx.doi.org/10.1016/S0091-679X(10)96028-7
- H. Fernández‐Morán, "LOW‐TEMPERATURE PREPARATION TECHNIQUES FOR ELECTRON MICROSCOPY OF BIOLOGICAL SPECIMENS BASED ON RAPID FREEZING WITH LIQUID HELIUM II*", Annals of the New York Academy of Sciences, vol. 85, pp. 689-713, 1960. http://dx.doi.org/10.1111/j.1749-6632.1960.tb49990.x
- M.T. Nicolas, J.M. Bassot, and G. Nicolas, "Immunogold labeling of luciferase in the luminous bacterium Vibrio harveyi after fast-freeze fixation and different freeze-substitution and embedding procedures.", Journal of Histochemistry & Cytochemistry, vol. 37, pp. 663-674, 1989. http://dx.doi.org/10.1177/37.5.2703702
- H. Schwarz, and B.M. Humbel, "Influence of fixatives and embedding media on immunolabelling of freeze-substituted cells.", Scanning microscopy. Supplement, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2694274
- K. McDONALD, and R. WEBB, "Freeze substitution in 3 hours or less", Journal of Microscopy, vol. 243, pp. 227-233, 2011. http://dx.doi.org/10.1111/j.1365-2818.2011.03526.x
- E. Behrman, "The chemistry of osmium tetroxide fixation", The Science and Biology of Specimen Preparation for Microscopy and Microanalysis 1-5., 1983.
- T.H. Giddings, "Freeze‐substitution protocols for improved visualization of membranes in high‐pressure frozen samples", Journal of Microscopy, vol. 212, pp. 53-61, 2003. http://dx.doi.org/10.1046/j.1365-2818.2003.01228.x
- O. Torres-Fernández, and N. Ordóñez, "Comparative effect of osmium tetroxide and ruthenium tetroxide on Penicillium sp. hyphae and Saccharomyces cerevisiae fungal cell wall ultrastructure.", Biomedica : revista del Instituto Nacional de Salud, 2003. http://www.ncbi.nlm.nih.gov/pubmed/12872562
- M. Osumi, "High-pressure freezing is a powerful tool for visualization of Schizosaccharomyces pombe cells: ultra-low temperature and low-voltage scanning electron microscopy and immunoelectron microscopy", Journal of Electron Microscopy, vol. 55, pp. 75-88, 2006. http://dx.doi.org/10.1093/jmicro/dfl014
- J.H. Luft, "IMPROVEMENTS IN EPOXY RESIN EMBEDDING METHODS", The Journal of Cell Biology, vol. 9, pp. 409-414, 1961. http://dx.doi.org/10.1083/jcb.9.2.409
- A.M. GLAUERT, G.E. ROGERS, and R.H. GLAUERT, "A New Embedding Medium for Electron Microscopy", Nature, vol. 178, pp. 803-803, 1956. http://dx.doi.org/10.1038/178803a0
- A. Bertin, M.A. McMurray, J. Pierson, L. Thai, K.L. McDonald, E.A. Zehr, G. García, P. Peters, J. Thorner, and E. Nogales, "Three-dimensional ultrastructure of the septin filament network inSaccharomyces cerevisiae", Molecular Biology of the Cell, vol. 23, pp. 423-432, 2012. http://dx.doi.org/10.1091/mbc.E11-10-0850
- P. Roberts, S. Moshitch-Moshkovitz, E. Kvam, E. O'Toole, M. Winey, and D.S. Goldfarb, "Piecemeal Microautophagy of Nucleus inSaccharomyces cerevisiae", Molecular Biology of the Cell, vol. 14, pp. 129-141, 2003. http://dx.doi.org/10.1091/mbc.E02-08-0483
- A. Prinz, E. Hartmann, and K. Kalies, "Sec61p Is the Main Ribosome Receptor in the Endoplasmic Reticulum of Saccharomyces cerevisiae", Biological Chemistry, vol. 381, pp. 1025-1028, 2000. http://dx.doi.org/10.1515/BC.2000.126
- M.N. Morin-Ganet, A. Rambourg, Y. Clermont, and F. Képès, "Role of endoplasmic reticulum-derived vesicles in the formation of Golgi elements in sec23 and sec18 Saccharomyces Cerevisiae mutants.", The Anatomical record, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9624457
- A.R. Spurr, "A low-viscosity epoxy resin embedding medium for electron microscopy", Journal of Ultrastructure Research, vol. 26, pp. 31-43, 1969. http://dx.doi.org/10.1016/S0022-5320(69)90033-1
- G.J. Hermann, J.W. Thatcher, J.P. Mills, K.G. Hales, M.T. Fuller, J. Nunnari, and J.M. Shaw, "Mitochondrial Fusion in Yeast Requires the Transmembrane GTPase Fzo1p", The Journal of Cell Biology, vol. 143, pp. 359-373, 1998. http://dx.doi.org/10.1083/jcb.143.2.359
- W. Bleazard, J.M. McCaffery, E.J. King, S. Bale, A. Mozdy, Q. Tieu, J. Nunnari, and J.M. Shaw, "The dynamin-related GTPase Dnm1 regulates mitochondrial fission in yeast", Nature Cell Biology, vol. 1, pp. 298-304, 1999. http://dx.doi.org/10.1038/13014
- H. Cheong, T. Yorimitsu, F. Reggiori, J.E. Legakis, C. Wang, and D.J. Klionsky, "Atg17 Regulates the Magnitude of the Autophagic Response", Molecular Biology of the Cell, vol. 16, pp. 3438-3453, 2005. http://dx.doi.org/10.1091/mbc.e04-10-0894
- M. Sekiya-Kawasaki, A.C. Groen, M.J.T. Cope, M. Kaksonen, H.A. Watson, C. Zhang, K.M. Shokat, B. Wendland, K.L. McDonald, J.M. McCaffery, and D.G. Drubin, "Dynamic phosphoregulation of the cortical actin cytoskeleton and endocytic machinery revealed by real-time chemical genetic analysis", The Journal of Cell Biology, vol. 162, pp. 765-772, 2003. http://dx.doi.org/10.1083/jcb.200305077
- K. Saito, R. Ohtomo, Y. Kuga-Uetake, T. Aono, and M. Saito, "Direct Labeling of Polyphosphate at the Ultrastructural Level inSaccharomyces cerevisiaeby Using the Affinity of the Polyphosphate Binding Domain ofEscherichia coliExopolyphosphatase", Applied and Environmental Microbiology, vol. 71, pp. 5692-5701, 2005. http://dx.doi.org/10.1128/aem.71.10.5692-5701.2005
- X. Pan, P. Roberts, Y. Chen, E. Kvam, N. Shulga, K. Huang, S. Lemmon, and D.S. Goldfarb, "Nucleus–Vacuole Junctions inSaccharomyces cerevisiaeAre Formed Through the Direct Interaction of Vac8p with Nvj1p", Molecular Biology of the Cell, vol. 11, pp. 2445-2457, 2000. http://dx.doi.org/10.1091/mbc.11.7.2445
- T. Darsow, S.E. Rieder, and S.D. Emr, "A Multispecificity Syntaxin Homologue, Vam3p, Essential for Autophagic and Biosynthetic Protein Transport to the Vacuole", The Journal of Cell Biology, vol. 138, pp. 517-529, 1997. http://dx.doi.org/10.1083/jcb.138.3.517
- R. Wright, M. Basson, L. D'Ari, and J. Rine, "Increased amounts of HMG-CoA reductase induce "karmellae": a proliferation of stacked membrane pairs surrounding the yeast nucleus.", The Journal of cell biology, vol. 107, pp. 101-114, 1988. http://dx.doi.org/10.1083/jcb.107.1.101
- S.E. Rieder, L.M. Banta, K. Köhrer, J.M. McCaffery, and S.D. Emr, "Multilamellar endosome-like compartment accumulates in the yeast vps28 vacuolar protein sorting mutant.", Molecular Biology of the Cell, vol. 7, pp. 985-999, 1996. http://dx.doi.org/10.1091/mbc.7.6.985
- R.C.N. Melo, E. Morgan, R. Monahan-Earley, A.M. Dvorak, and P.F. Weller, "Pre-embedding immunogold labeling to optimize protein localization at subcellular compartments and membrane microdomains of leukocytes", Nature Protocols, vol. 9, pp. 2382-2394, 2014. http://dx.doi.org/10.1038/nprot.2014.163
- J. Acetarin, E. Carlemalm, and W. Villiger, "Developments of new Lowicryl® resins for embedding biological specimens at even lower temperatures", Journal of Microscopy, vol. 143, pp. 81-88, 1986. http://dx.doi.org/10.1111/j.1365-2818.1986.tb02766.x
- G.A. Petsko, "Protein crystallography at sub-zero temperatures: Cryo-protective mother liquors for protein crystals", Journal of Molecular Biology, vol. 96, pp. 381-392, 1975. http://dx.doi.org/10.1016/0022-2836(75)90167-9
- E. Kellenberger, M. Dürrenberger, W. Villiger, E. Carlemalm, and M. Wurtz, "The efficiency of immunolabel on Lowicryl sections compared to theoretical predictions.", Journal of Histochemistry & Cytochemistry, vol. 35, pp. 959-969, 1987. http://dx.doi.org/10.1177/35.9.3302020
- C. Weibull, A. Christiansson, and E. Carlemalm, "Extraction of membrane lipids during fixation, dehydration and embedding of Acholeplasma laidlawii‐cells for electron microscopy", Journal of Microscopy, vol. 129, pp. 201-207, 1983. http://dx.doi.org/10.1111/j.1365-2818.1983.tb04174.x
- C.M.J. Sagt, W.H. Müller, L. van der Heide, J. Boonstra, A.J. Verkleij, and C.T. Verrips, "Impaired Cutinase Secretion in Saccharomyces cerevisiae Induces Irregular Endoplasmic Reticulum (ER) Membrane Proliferation, Oxidative Stress, and ER-Associated Degradation", Applied and Environmental Microbiology, vol. 68, pp. 2155-2160, 2002. http://dx.doi.org/10.1128/AEM.68.5.2155-2160.2002
- B.E. Causton, "Improved bonding of composite restorative to dentine. A study in vitro of the use of a commercial halogenated phosphate ester", British Dental Journal, vol. 156, pp. 93-95, 1984. http://dx.doi.org/10.1038/sj.bdj.4805276
- J.P. Aris, and G. Blobel, "Yeast nuclear envelope proteins cross react with an antibody against mammalian pore complex proteins.", The Journal of cell biology, vol. 108, pp. 2059-2067, 1989. http://dx.doi.org/10.1083/jcb.108.6.2059
- L. Popolo, M. Vai, E. Gatti, S. Porello, P. Bonfante, R. Balestrini, and L. Alberghina, "Physiological analysis of mutants indicates involvement of the Saccharomyces cerevisiae GPI-anchored protein gp115 in morphogenesis and cell separation.", Journal of bacteriology, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8458831
- C. Prescianotto-Baschong, and H. Riezman, "Morphology of the Yeast Endocytic Pathway", Molecular Biology of the Cell, vol. 9, pp. 173-189, 1998. http://dx.doi.org/10.1091/mbc.9.1.173
- C. Prescianotto‐Baschong, and H. Riezman, "Ordering of Compartments in the Yeast Endocytic Pathway", Traffic, vol. 3, pp. 37-49, 2002. http://dx.doi.org/10.1034/j.1600-0854.2002.30106.x
- J. Mulholland, J. Konopka, B. Singer-Kruger, M. Zerial, and D. Botstein, "Visualization of Receptor-mediated Endocytosis in Yeast", Molecular Biology of the Cell, vol. 10, pp. 799-817, 1999. http://dx.doi.org/10.1091/mbc.10.3.799
- J. Mulholland, D. Preuss, A. Moon, A. Wong, D. Drubin, and D. Botstein, "Ultrastructure of the yeast actin cytoskeleton and its association with the plasma membrane.", The Journal of cell biology, vol. 125, pp. 381-391, 1994. http://dx.doi.org/10.1083/jcb.125.2.381
- S. Bernales, K.L. McDonald, and P. Walter, "Autophagy Counterbalances Endoplasmic Reticulum Expansion during the Unfolded Protein Response", PLoS Biology, vol. 4, pp. e423, 2006. http://dx.doi.org/10.1371/journal.pbio.0040423
- V.J. Cid, M.J. Shulewitz, K.L. McDonald, and J. Thorner, "Dynamic Localization of the Swe1 Regulator Hsl7 During theSaccharomyces cerevisiaeCell Cycle", Molecular Biology of the Cell, vol. 12, pp. 1645-1669, 2001. http://dx.doi.org/10.1091/mbc.12.6.1645
- K. Tokuyasu, "A study of positive staining of ultrathin frozen sections", Journal of Ultrastructure Research, vol. 63, pp. 287-307, 1978. http://dx.doi.org/10.1016/S0022-5320(78)80053-7
- K.T. Tokuyasu, "A TECHNIQUE FOR ULTRACRYOTOMY OF CELL SUSPENSIONS AND TISSUES", The Journal of Cell Biology, vol. 57, pp. 551-565, 1973. http://dx.doi.org/10.1083/jcb.57.2.551
- K. Cortese, A. Diaspro, and C. Tacchetti, "Advanced Correlative Light/Electron Microscopy: Current Methods and New Developments Using Tokuyasu Cryosections", Journal of Histochemistry & Cytochemistry, vol. 57, pp. 1103-1112, 2009. http://dx.doi.org/10.1369/jhc.2009.954214
- J. Griffith, M. Mari, A. De Mazière, and F. Reggiori, "A Cryosectioning Procedure for the Ultrastructural Analysis and the Immunogold Labelling of Yeast Saccharomyces cerevisiae", Traffic, vol. 9, pp. 1060-1072, 2008. http://dx.doi.org/10.1111/j.1600-0854.2008.00753.x
- C. Hönscher, M. Mari, K. Auffarth, M. Bohnert, J. Griffith, W. Geerts, M. van der Laan, M. Cabrera, F. Reggiori, and C. Ungermann, "Cellular Metabolism Regulates Contact Sites between Vacuoles and Mitochondria", Developmental Cell, vol. 30, pp. 86-94, 2014. http://dx.doi.org/10.1016/j.devcel.2014.06.006
- M. Harner, C. Körner, D. Walther, D. Mokranjac, J. Kaesmacher, U. Welsch, J. Griffith, M. Mann, F. Reggiori, and W. Neupert, "The mitochondrial contact site complex, a determinant of mitochondrial architecture", The EMBO Journal, vol. 30, pp. 4356-4370, 2011. http://dx.doi.org/10.1038/emboj.2011.379
- M. Cabrera, L. Langemeyer, M. Mari, R. Rethmeier, I. Orban, A. Perz, C. Bröcker, J. Griffith, D. Klose, H. Steinhoff, F. Reggiori, S. Engelbrecht-Vandré, and C. Ungermann, "Phosphorylation of a membrane curvature–sensing motif switches function of the HOPS subunit Vps41 in membrane tethering", Journal of Cell Biology, vol. 191, pp. 845-859, 2010. http://dx.doi.org/10.1083/jcb.201004092
- D.F. Markgraf, F. Ahnert, H. Arlt, M. Mari, K. Peplowska, N. Epp, J. Griffith, F. Reggiori, and C. Ungermann, "The CORVET Subunit Vps8 Cooperates with the Rab5 Homolog Vps21 to Induce Clustering of Late Endosomal Compartments", Molecular Biology of the Cell, vol. 20, pp. 5276-5289, 2009. http://dx.doi.org/10.1091/mbc.E09-06-0521
- K.E. Moreira, S. Schuck, B. Schrul, F. Fröhlich, J.B. Moseley, T.C. Walther, and P. Walter, "Seg1 controls eisosome assembly and shape", Journal of Cell Biology, vol. 198, pp. 405-420, 2012. http://dx.doi.org/10.1083/jcb.201202097
- A. Chadrin, B. Hess, M. San Roman, X. Gatti, B. Lombard, D. Loew, Y. Barral, B. Palancade, and V. Doye, "Pom33, a novel transmembrane nucleoporin required for proper nuclear pore complex distribution", Journal of Cell Biology, vol. 189, pp. 795-811, 2010. http://dx.doi.org/10.1083/jcb.200910043
- J. Sawa-Makarska, C. Abert, J. Romanov, B. Zens, I. Ibiricu, and S. Martens, "Cargo binding to Atg19 unmasks additional Atg8 binding sites to mediate membrane–cargo apposition during selective autophagy", Nature Cell Biology, vol. 16, pp. 425-433, 2014. http://dx.doi.org/10.1038/ncb2935
- U. Nair, A. Jotwani, J. Geng, N. Gammoh, D. Richerson, W. Yen, J. Griffith, S. Nag, K. Wang, T. Moss, M. Baba, J. McNew, X. Jiang, F. Reggiori, T. Melia, and D. Klionsky, "SNARE Proteins Are Required for Macroautophagy", Cell, vol. 146, pp. 290-302, 2011. http://dx.doi.org/10.1016/j.cell.2011.06.022
- M. Mari, J. Griffith, and F. Reggiori, "Investigation of the ultrastructure of yeast using cryo-immuno-electron microscopy", Microscopy: Science, Technology, Applications and Education 1(4): 11-18., 2010.
- E. van Donselaar, G. Posthuma, D. Zeuschner, B.M. Humbel, and J.W. Slot, "Immunogold Labeling of Cryosections from High‐Pressure Frozen Cells", Traffic, vol. 8, pp. 471-485, 2007. http://dx.doi.org/10.1111/j.1600-0854.2007.00552.x
- B.A. Afzelius, "Section staining for electron microscopy using tannic acid as a mordant: A simple method for visualization of glycogen and collagen", Microscopy Research and Technique, vol. 21, pp. 65-72, 1992. http://dx.doi.org/10.1002/jemt.1070210110
- J.M. Frasca, and V.R. Parks, "A ROUTINE TECHNIQUE FOR DOUBLE-STAINING ULTRATHIN SECTIONS USING URANYL AND LEAD SALTS", The Journal of Cell Biology, vol. 25, pp. 157-161, 1965. http://dx.doi.org/10.1083/jcb.25.1.157
- T. Sato, "A modified method for lead staining of thin sections.", Journal of electron microscopy, 1968. http://www.ncbi.nlm.nih.gov/pubmed/4177281
- R.W. Horne, "The Effects of Negative Stains on Lipids and Proteins Observed in the Electron Microscope", Symposium on Biophysics and Physiology of Biological Transport, pp. 212-213, 1967. http://dx.doi.org/10.1007/978-3-662-25134-8_49
- Y. Namiki, K. Ueno, H. Mitani, E.V. Virtudazo, M. Ohkusu, K. Shimizu, S. Kawamoto, H. Chibana, and M. Yamaguchi, "Scanning and negative-staining electron microscopy of protoplast regeneration of a wild-type and two chitin synthase mutants in the pathogenic yeast Candida glabrata", Journal of Electron Microscopy, vol. 60, pp. 157-165, 2011. http://dx.doi.org/10.1093/jmicro/dfq082
- V. Busygina, W.A. Gaines, Y. Xu, Y. Kwon, G.J. Williams, S. Lin, H. Chang, P. Chi, H. Wang, and P. Sung, "Functional attributes of the Saccharomyces cerevisiae meiotic recombinase Dmc1", DNA Repair, vol. 12, pp. 707-712, 2013. http://dx.doi.org/10.1016/j.dnarep.2013.05.004
- B. Mizuhira, and T. Amakawa, "[Electron microscopic studies on the histocytochemistry and the physiological function of the kidney].", Nihon rinsho. Japanese journal of clinical medicine, 1972. http://www.ncbi.nlm.nih.gov/pubmed/4262981
- J.V. Kilmartin, "Purification of yeast tubulin by self-assembly in vitro", Biochemistry, vol. 20, pp. 3629-3633, 1981. http://dx.doi.org/10.1021/bi00515a050
- E. Kiseleva, A.C. Richardson, J. Fiserova, A.A. Strunov, M.C. Spink, S.R. Johnson, and M.W. Goldberg, "Imaging Yeast NPCs", Methods in Cell Biology, pp. 59-79, 2014. http://dx.doi.org/10.1016/B978-0-12-417160-2.00003-5
- Y. Shimoni, T. Kurihara, M. Ravazzola, M. Amherdt, L. Orci, and R. Schekman, "Lst1p and Sec24p Cooperate in Sorting of the Plasma Membrane Atpase into Copii Vesicles in Saccharomyces cerevisiae", The Journal of Cell Biology, vol. 151, pp. 973-984, 2000. http://dx.doi.org/10.1083/jcb.151.5.973
- S.M. Wilson, and A. Bacic, "Preparation of plant cells for transmission electron microscopy to optimize immunogold labeling of carbohydrate and protein epitopes", Nature Protocols, vol. 7, pp. 1716-1727, 2012. http://dx.doi.org/10.1038/nprot.2012.096
- T.G. Frey, G.A. Perkins, and M.H. Ellisman, "ELECTRON TOMOGRAPHY OF MEMBRANE-BOUND CELLULAR ORGANELLES", Annual Review of Biophysics and Biomolecular Structure, vol. 35, pp. 199-224, 2006. http://dx.doi.org/10.1146/annurev.biophys.35.040405.102039
- A. Perktold, B. Zechmann, G. Daum, and G. Zellnig, "Organelle association visualized by three-dimensional ultrastructural imaging of the yeast cell", FEMS Yeast Research, vol. 7, pp. 629-638, 2007. http://dx.doi.org/10.1111/j.1567-1364.2007.00226.x
- M. Yamaguchi, H. Okada, and Y. Namiki, "Smart specimen preparation for freeze substitution and serial ultrathin sectioning of yeast cells", Journal of Electron Microscopy, vol. 58, pp. 261-266, 2009. http://dx.doi.org/10.1093/jmicro/dfp013
- M. Osumi, "The ultrastructure of yeast: Cell wall structure and formation", Micron, vol. 29, pp. 207-233, 1998. http://dx.doi.org/10.1016/s0968-4328(97)00072-3
- M. Yamaguchi, Y. Namiki, H. Okada, Y. Mori, H. Furukawa, J. Wang, M. Ohkusu, and S. Kawamoto, "Structome of Saccharomyces cerevisiae determined by freeze-substitution and serial ultrathin-sectioning electron microscopy", Microscopy, vol. 60, pp. 321-335, 2011. http://dx.doi.org/10.1093/jmicro/dfr052
- H. Pichler, B. Gaigg, C. Hrastnik, G. Achleitner, S.D. Kohlwein, G. Zellnig, A. Perktold, and G. Daum, "A subfraction of the yeast endoplasmic reticulum associates with the plasma membrane and has a high capacity to synthesize lipids", European Journal of Biochemistry, vol. 268, pp. 2351-2361, 2001. http://dx.doi.org/10.1046/j.1432-1327.2001.02116.x
- G. Achleitner, B. Gaigg, A. Krasser, E. Kainersdorfer, S.D. Kohlwein, A. Perktold, G. Zellnig, and G. Daum, "Association between the endoplasmic reticulum and mitochondria of yeast facilitates interorganelle transport of phospholipids through membrane contact", European Journal of Biochemistry, vol. 264, pp. 545-553, 1999. http://dx.doi.org/10.1046/j.1432-1327.1999.00658.x
- V. Lučić, F. Förster, and W. Baumeister, "STRUCTURAL STUDIES BY ELECTRON TOMOGRAPHY: From Cells to Molecules", Annual Review of Biochemistry, vol. 74, pp. 833-865, 2005. http://dx.doi.org/10.1146/annurev.biochem.73.011303.074112
- E.T. O'Toole, M. Winey, J. McIntosh, and D.N. Mastronarde, "Electron tomography of yeast cells", Guide to Yeast Genetics and Molecular and Cell Biology Part C, pp. 81-96, 2002. http://dx.doi.org/10.1016/S0076-6879(02)51842-5
- M. Winey, C.L. Mamay, E.T. O'Toole, D.N. Mastronarde, T.H. Giddings, K.L. McDonald, and J.R. McIntosh, "Three-dimensional ultrastructural analysis of the Saccharomyces cerevisiae mitotic spindle.", The Journal of cell biology, vol. 129, pp. 1601-1615, 1995. http://dx.doi.org/10.1083/jcb.129.6.1601
- E. Nazarova, E. O'Toole, S. Kaitna, P. Francois, M. Winey, and J. Vogel, "Distinct roles for antiparallel microtubule pairing and overlap during early spindle assembly", Molecular Biology of the Cell, vol. 24, pp. 3238-3250, 2013. http://dx.doi.org/10.1091/mbc.E13-05-0232
- P. Melloy, S. Shen, E. White, J.R. McIntosh, and M.D. Rose, "Nuclear fusion during yeast mating occurs by a three-step pathway", The Journal of Cell Biology, vol. 179, pp. 659-670, 2007. http://dx.doi.org/10.1083/jcb.200706151
- D.P. Nickerson, M. West, and G. Odorizzi, "Did2 coordinates Vps4-mediated dissociation of ESCRT-III from endosomes", The Journal of Cell Biology, vol. 175, pp. 715-720, 2006. http://dx.doi.org/10.1083/jcb.200606113
- M.W. McNatt, I. McKittrick, M. West, and G. Odorizzi, "Direct Binding to Rsp5 Mediates Ubiquitin-independent Sorting of Sna3 via the Multivesicular Body Pathway", Molecular Biology of the Cell, vol. 18, pp. 697-706, 2007. http://dx.doi.org/10.1091/mbc.e06-08-0663
- C. Richter, M. West, and G. Odorizzi, "Dual mechanisms specify Doa4-mediated deubiquitination at multivesicular bodies", The EMBO Journal, vol. 26, pp. 2454-2464, 2007. http://dx.doi.org/10.1038/sj.emboj.7601692
- J.R. Friedman, L.L. Lackner, M. West, J.R. DiBenedetto, J. Nunnari, and G.K. Voeltz, "ER Tubules Mark Sites of Mitochondrial Division", Science, vol. 334, pp. 358-362, 2011. http://dx.doi.org/10.1126/science.1207385
- S. Hoppins, S.R. Collins, A. Cassidy-Stone, E. Hummel, R.M. DeVay, L.L. Lackner, B. Westermann, M. Schuldiner, J.S. Weissman, and J. Nunnari, "A mitochondrial-focused genetic interaction map reveals a scaffold-like complex required for inner membrane organization in mitochondria", Journal of Cell Biology, vol. 195, pp. 323-340, 2011. http://dx.doi.org/10.1083/jcb.201107053
- A. Murley, L.L. Lackner, C. Osman, M. West, G.K. Voeltz, P. Walter, and J. Nunnari, "ER-associated mitochondrial division links the distribution of mitochondria and mitochondrial DNA in yeast", eLife, vol. 2, 2013. http://dx.doi.org/10.7554/eLife.00422
- W. Kukulski, M. Schorb, M. Kaksonen, and J. Briggs, "Plasma Membrane Reshaping during Endocytosis Is Revealed by Time-Resolved Electron Tomography", Cell, vol. 150, pp. 508-520, 2012. http://dx.doi.org/10.1016/j.cell.2012.05.046
- H. Wolinski, D. Kolb, S. Hermann, R.I. Koning, and S.D. Kohlwein, "A role for seipin in lipid droplet dynamics and inheritance in yeast", Journal of Cell Science, vol. 124, pp. 3894-3904, 2011. http://dx.doi.org/10.1242/jcs.091454
- E. Sokol, D. Kramer, G.F. Diercks, J. Kuipers, M.F. Jonkman, H.H. Pas, and B.N. Giepmans, "Large-Scale Electron Microscopy Maps of Patient Skin and Mucosa Provide Insight into Pathogenesis of Blistering Diseases", Journal of Investigative Dermatology, vol. 135, pp. 1763-1770, 2015. http://dx.doi.org/10.1038/jid.2015.109
- W.H. Escovitz, T.R. Fox, and R. Levi-Setti, "Scanning transmission ion microscope with a field ion source.", Proceedings of the National Academy of Sciences, vol. 72, pp. 1826-1828, 1975. http://dx.doi.org/10.1073/pnas.72.5.1826
- C. Villinger, H. Gregorius, C. Kranz, K. Höhn, C. Münzberg, G. Wichert, B. Mizaikoff, G. Wanner, and P. Walther, "FIB/SEM tomography with TEM-like resolution for 3D imaging of high-pressure frozen cells", Histochemistry and Cell Biology, vol. 138, pp. 549-556, 2012. http://dx.doi.org/10.1007/s00418-012-1020-6
- D. Wei, S. Jacobs, S. Modla, S. Zhang, C.L. Young, R. Cirino, J. Caplan, and K. Czymmek, "High-resolution three-dimensional reconstruction of a whole yeast cell using focused-ion beam scanning electron microscopy.", BioTechniques, 2012. http://www.ncbi.nlm.nih.gov/pubmed/22780318
- N. Miyazaki, M. Esaki, T. Ogura, and K. Murata, "Serial block-face scanning electron microscopy for three-dimensional analysis of morphological changes in mitochondria regulated by Cdc48p/p97 ATPase", Journal of Structural Biology, vol. 187, pp. 187-193, 2014. http://dx.doi.org/10.1016/j.jsb.2014.05.010
- P. Ngamskulrungroj, Y. Chang, B. Hansen, C. Bugge, E. Fischer, and K.J. Kwon-Chung, "Characterization of the Chromosome 4 Genes That Affect Fluconazole-Induced Disomy Formation in Cryptococcus neoformans", PLoS ONE, vol. 7, pp. e33022, 2012. http://dx.doi.org/10.1371/journal.pone.0033022
- M.F. Hayles, D. Matthijs de Winter, C.T. Schneijdenberg, J.D. Meeldijk, U. Luecken, H. Persoon, J. de Water, F. de Jong, B.M. Humbel, and A.J. Verkleij, "The making of frozen-hydrated, vitreous lamellas from cells for cryo-electron microscopy", Journal of Structural Biology, vol. 172, pp. 180-190, 2010. http://dx.doi.org/10.1016/j.jsb.2010.07.004
-
T. Bo, M. Liu, C. Zhong, Q. Zhang, Q. Su, Z. Tan, P. Han, and S. Jia, "Metabolomic Analysis of Antimicrobial Mechanisms of ε-Poly-
l -lysine on Saccharomyces cerevisiae", Journal of Agricultural and Food Chemistry, vol. 62, pp. 4454-4465, 2014. http://dx.doi.org/10.1021/jf500505n - S. Bennis, F. Chami, N. Chami, T. Bouchikhi, and A. Remmal, "Surface alteration of Saccharomyces cerevisiae induced by thymol and eugenol", Letters in Applied Microbiology, vol. 38, pp. 454-458, 2004. http://dx.doi.org/10.1111/j.1472-765X.2004.01511.x
- A. Coluccio, "Interspore bridges: a new feature of the Saccharomyces cerevisiae spore wall", Microbiology, vol. 150, pp. 3189-3196, 2004. http://dx.doi.org/10.1099/mic.0.27253-0
- C. Ferreira, S. Silva, F. van Voorst, C. Aguiar, M.C. Kielland-Brandt, A. Brandt, and C. Lucas, "Absence of Gup1p inSaccharomyces cerevisiaeresults in defective cell wall composition, assembly, stability and morphology", FEMS Yeast Research, vol. 6, pp. 1027-1038, 2006. http://dx.doi.org/10.1111/j.1567-1364.2006.00110.x
- F.J. Timmermans, and C. Otto, "Contributed Review: Review of integrated correlative light and electron microscopy", Review of Scientific Instruments, vol. 86, 2015. http://dx.doi.org/10.1063/1.4905434
- P. de Boer, J.P. Hoogenboom, and B.N.G. Giepmans, "Correlated light and electron microscopy: ultrastructure lights up!", Nature Methods, vol. 12, pp. 503-513, 2015. http://dx.doi.org/10.1038/nmeth.3400
- C. Loussert Fonta, and B.M. Humbel, "Correlative microscopy", Archives of Biochemistry and Biophysics, vol. 581, pp. 98-110, 2015. http://dx.doi.org/10.1016/j.abb.2015.05.017
- R.T. Schirra, and P. Zhang, "Correlative Fluorescence and Electron Microscopy", Current Protocols in Cytometry, vol. 70, 2014. http://dx.doi.org/10.1002/0471142956.cy1236s70
- C. Buser, and K. McDonald, "Correlative GFP-Immunoelectron Microscopy in Yeast", Methods in Enzymology, pp. 603-618, 2010. http://dx.doi.org/10.1016/S0076-6879(10)70024-0
- S. Viswanathan, M.E. Williams, E.B. Bloss, T.J. Stasevich, C.M. Speer, A. Nern, B.D. Pfeiffer, B.M. Hooks, W. Li, B.P. English, T. Tian, G.L. Henry, J.J. Macklin, R. Patel, C.R. Gerfen, X. Zhuang, Y. Wang, G.M. Rubin, and L.L. Looger, "High-performance probes for light and electron microscopy", Nature Methods, vol. 12, pp. 568-576, 2015. http://dx.doi.org/10.1038/nmeth.3365
- D.M. de Winter, R.J. Mesman, M.F. Hayles, C.T. Schneijdenberg, C. Mathisen, and J.A. Post, "In-situ integrity control of frozen-hydrated, vitreous lamellas prepared by the cryo-focused ion beam-scanning electron microscope", Journal of Structural Biology, vol. 183, pp. 11-18, 2013. http://dx.doi.org/10.1016/j.jsb.2013.05.016
- D. Studer, A. Klein, I. Iacovache, H. Gnaegi, and B. Zuber, "A new tool based on two micromanipulators facilitates the handling of ultrathin cryosection ribbons", Journal of Structural Biology, vol. 185, pp. 125-128, 2014. http://dx.doi.org/10.1016/j.jsb.2013.11.005
- . Walther, and . Müller, "Biological ultrastructure as revealed by high resolution cryo‐SEM of block faces after cryo‐sectioning", Journal of Microscopy, vol. 196, pp. 279-287, 1999. http://dx.doi.org/10.1046/j.1365-2818.1999.00595.x
- E. Bos, L. Hussaarts, J.R. van Weering, M.H. Ellisman, H. de Wit, and A.J. Koster, "Vitrification of Tokuyasu-style immuno-labelled sections for correlative cryo light microscopy and cryo electron tomography", Journal of Structural Biology, vol. 186, pp. 273-282, 2014. http://dx.doi.org/10.1016/j.jsb.2014.03.021
- S. Kawai-Noma, C. Pack, T. Kojidani, H. Asakawa, Y. Hiraoka, M. Kinjo, T. Haraguchi, H. Taguchi, and A. Hirata, "In vivo evidence for the fibrillar structures of Sup35 prions in yeast cells", Journal of Cell Biology, vol. 190, pp. 223-231, 2010. http://dx.doi.org/10.1083/jcb.201002149
- M. Michel, T. Hillmann, and M. Müller, "Cryosectioning of plant material frozen at high pressure", Journal of Microscopy, vol. 163, pp. 3-18, 1991. http://dx.doi.org/10.1111/j.1365-2818.1991.tb03155.x
- V.R.F. Matias, A. Al-Amoudi, J. Dubochet, and T.J. Beveridge, "Cryo-Transmission Electron Microscopy of Frozen-Hydrated Sections ofEscherichia coliandPseudomonas aeruginosa", Journal of Bacteriology, vol. 185, pp. 6112-6118, 2003. http://dx.doi.org/10.1128/jb.185.20.6112-6118.2003
- A. Al-Amoudi, J. Chang, A. Leforestier, A. McDowall, L.M. Salamin, L.P.O. Norlén, K. Richter, N.S. Blanc, D. Studer, and J. Dubochet, "Cryo-electron microscopy of vitreous sections", The EMBO Journal, vol. 23, pp. 3583-3588, 2004. http://dx.doi.org/10.1038/sj.emboj.7600366
- A. Al-Amoudi, L.P. Norlen, and J. Dubochet, "Cryo-electron microscopy of vitreous sections of native biological cells and tissues", Journal of Structural Biology, vol. 148, pp. 131-135, 2004. http://dx.doi.org/10.1016/j.jsb.2004.03.010
- A. Rigort, E. Villa, F.J. Bäuerlein, B.D. Engel, and J.M. Plitzko, "Integrative Approaches for Cellular Cryo-electron Tomography", Methods in Cell Biology, pp. 259-281, 2012. http://dx.doi.org/10.1016/B978-0-12-416026-2.00014-5
- C.A. Larabell, and M.A. Le Gros, "X-ray Tomography Generates 3-D Reconstructions of the Yeast, Saccharomyces cerevisiae, at 60-nm Resolution", Molecular Biology of the Cell, vol. 15, pp. 957-962, 2004. http://dx.doi.org/10.1091/mbc.E03-07-0522
- D. Weiß, G. Schneider, B. Niemann, P. Guttmann, D. Rudolph, and G. Schmahl, "Computed tomography of cryogenic biological specimens based on X-ray microscopic images", Ultramicroscopy, vol. 84, pp. 185-197, 2000. http://dx.doi.org/10.1016/S0304-3991(00)00034-6
- M.A. Le Gros, G. McDermott, and C.A. Larabell, "X-ray tomography of whole cells", Current Opinion in Structural Biology, vol. 15, pp. 593-600, 2005. http://dx.doi.org/10.1016/j.sbi.2005.08.008
- G. McDermott, M.A. Le Gros, C.G. Knoechel, M. Uchida, and C.A. Larabell, "Soft X-ray tomography and cryogenic light microscopy: the cool combination in cellular imaging", Trends in Cell Biology, vol. 19, pp. 587-595, 2009. http://dx.doi.org/10.1016/j.tcb.2009.08.005
- M. Uchida, G. McDermott, M. Wetzler, M.A. Le Gros, M. Myllys, C. Knoechel, A.E. Barron, and C.A. Larabell, "Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans", Proceedings of the National Academy of Sciences, vol. 106, pp. 19375-19380, 2009. http://dx.doi.org/10.1073/pnas.0906145106
- B.P. Cinquin, M. Do, G. McDermott, A.D. Walters, M. Myllys, E.A. Smith, O. Cohen‐Fix, M.A. Le Gros, and C.A. Larabell, "Putting Molecules in Their Place", Journal of Cellular Biochemistry, vol. 115, pp. 209-216, 2013. http://dx.doi.org/10.1002/jcb.24658
- T.L. Burnett, S.A. McDonald, A. Gholinia, R. Geurts, M. Janus, T. Slater, S.J. Haigh, C. Ornek, F. Almuaili, D.L. Engelberg, G.E. Thompson, and P.J. Withers, "Correlative Tomography", Scientific Reports, vol. 4, 2014. http://dx.doi.org/10.1038/srep04711
- M. Uchida, Y. Sun, G. McDermott, C. Knoechel, M.A. Le Gros, D. Parkinson, D.G. Drubin, and C.A. Larabell, "Quantitative analysis of yeast internal architecture using soft X‐ray tomography", Yeast, vol. 28, pp. 227-236, 2010. http://dx.doi.org/10.1002/yea.1834
- X. Huang, J. Nelson, J. Kirz, E. Lima, S. Marchesini, H. Miao, A.M. Neiman, D. Shapiro, J. Steinbrener, A. Stewart, J.J. Turner, and C. Jacobsen, "Soft X-Ray Diffraction Microscopy of a Frozen Hydrated Yeast Cell", Physical Review Letters, vol. 103, 2009. http://dx.doi.org/10.1103/physrevlett.103.198101
- A. Walters, C. May, E. Dauster, B. Cinquin, E. Smith, X. Robellet, D. D’Amours, C. Larabell, and O. Cohen-Fix, "The Yeast Polo Kinase Cdc5 Regulates the Shape of the Mitotic Nucleus", Current Biology, vol. 24, pp. 2861-2867, 2014. http://dx.doi.org/10.1016/j.cub.2014.10.029
- C. Howard, and M. Reed, "Unbiased stereology: Three-dimensional measurement in microscopy", 2nd ed. New York: Garland Science/BIOS Scientific Publishers., 2005.
- T.M. Mayhew, J.M. Lucocq, and G. Griffiths, "Relative labelling index: a novel stereological approach to test for non‐random immunogold labelling of organelles and membranes on transmission electron microscopy thin sections", Journal of Microscopy, vol. 205, pp. 153-164, 2002. http://dx.doi.org/10.1046/j.0022-2720.2001.00977.x
- C. Rabouille, "Quantitative Aspects of Immunogold Labeling in Embedded and Nonembedded Sections", Electron Microscopy Methods and Protocols, pp. 125-144, . http://dx.doi.org/10.1385/1-59259-201-5:125
- M.S. Longtine, A. McKenzie, D.J. Demarini, N.G. Shah, A. Wach, A. Brachat, P. Philippsen, and J.R. Pringle, "Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae.", Yeast (Chichester, England), 1998. http://www.ncbi.nlm.nih.gov/pubmed/9717241
- M. Binder, A. Hartig, and T. Sata, "Immunogold labeling of yeast cells: an efficient tool for the study of protein targeting and morphological alterations due to overexpression and inactivation of genes", Histochemistry and Cell Biology, vol. 106, pp. 115-130, 1996. http://dx.doi.org/10.1007/BF02473206
- M. Cabrera, H. Arlt, N. Epp, J. Lachmann, J. Griffith, A. Perz, F. Reggiori, and C. Ungermann, "Functional Separation of Endosomal Fusion Factors and the Class C Core Vacuole/Endosome Tethering (CORVET) Complex in Endosome Biogenesis", Journal of Biological Chemistry, vol. 288, pp. 5166-5175, 2013. http://dx.doi.org/10.1074/jbc.M112.431536
ACKNOWLEDGMENTS
The authors apologize to those researchers whose work is not included or cited due to space limitations. The authors thank Willie Geerts, Ben Giepmans and Kevin Knoops for the critical reading of the manuscript. F.R. is supported by ALW Open Program (822.02.014), DFG-NWO cooperation (DN82-303), SNF Sinergia (CRSII3_154421) and ZonMW VICI (016.130.606) grants.
COPYRIGHT
© 2015

Electron microscopy for ultrastructural analysis and protein localization in Saccharomyces cerevisiae by Andri Frankl et al. is licensed under a Creative Commons Attribution 4.0 International License.








