
Research Articles:
Microbial Cell, Vol. 1, No. 1, pp. 21 - 36; doi: 10.15698/mic2014.01.121
Reduced TORC1 signaling abolishes mitochondrial dysfunctions and shortened chronological lifespan of Isc1p-deficient cells
1 Instituto de Biologia Molecular e Celular, Rua do Campo Alegre, 823, 4150-180 Porto, Portugal
2 Departamento de Biologia Molecular, Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
Keywords: Isc1p; Tor1p; Sch9p; oxidative stress; chronological aging, mitochondrial function, ROS, ceramide.
Received originally: 06/11/2013 Received in revised form: 05/12/2013
Accepted: 11/12/2013
Published: 06/01/2014
Correspondence:
Vítor Costa, Instituto de Biologia Molecular e Celular, Rua do Campo Alegre, 823; 4150-180 Porto, Portugal. vcosta@ibmc.up.pt
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Vitor Teixeira, Tânia C. Medeiros, Rita Vilaça, Pedro Moradas-Ferreira, and Vítor Costa (2014). Reduced TORC1 signaling abolishes mitochondrial dysfunctions and shortened chronological lifespan of Isc1p-deficient cells. Microbial Cell 1(1): 21-36.
Abstract
The target of rapamycin (TOR) is an important signaling pathway on a hierarchical network of interacting pathways regulating central biological processes, such as cell growth, stress response and aging. Several lines of evidence suggest a functional link between TOR signaling and sphingolipid metabolism. Here, we report that the TORC1-Sch9p pathway is activated in cells lacking Isc1p, the yeast orthologue of mammalian neutral sphingomyelinase 2. The deletion of TOR1 or SCH9 abolishes the premature aging, oxidative stress sensitivity and mitochondrial dysfunctions displayed by isc1Δ cells and this is correlated with the suppression of the autophagic flux defect exhibited by the mutant strain. The protective effect of TOR1 deletion, as opposed to that of SCH9 deletion, is not associated with the attenuation of Hog1p hyperphosphorylation, which was previously implicated in isc1Δ phenotypes. Our data support a model in which Isc1p regulates mitochondrial function and chronological lifespan in yeast through the TORC1-Sch9p pathway although Isc1p and TORC1 also seem to act through independent pathways, as isc1Δtor1Δ phenotypes are intermediate to those displayed by isc1Δ and tor1Δ cells. We also provide evidence that TORC1 downstream effectors, the type 2A protein phosphatase Sit4p and the AGC protein kinase Sch9p, integrate nutrient and stress signals from TORC1 with ceramide signaling derived from Isc1p to regulate mitochondrial function and lifespan in yeast. Overall, our results show that TORC1-Sch9p axis is deregulated in Isc1p-deficient cells, contributing to mitochondrial dysfunction, enhanced oxidative stress sensitivity and premature aging of isc1Δ cells.
INTRODUCTION
Sphingolipids are ubiquitous structural components of eukaryotic cell membranes, and their bioactive metabolites (sphingosine, sphingosine-1-phosphate, ceramide, ceramide-1‑phosphate and lyso-sphingomyelin) are known to act as second messengers in the regulation of signaling pathways [1],[2],[3],[4],[5],[6]. Sphingosine (and related sphingoid bases) and ceramide are involved in the regulation of actin cytoskeleton organization, endocytosis, degradation of nutrient permeases, apoptosis, cell senescence and cell cycle arrest whereas sphingosine-1-phosphate plays a key role in proliferation, mitogenesis, cell migration, cell survival and inflammation (in higher eukaryotes) [5]. Thus, subtle variations on the relative amounts of sphingosine-1-phosphate and sphingosine/ceramide are expected to determine cell fate in response to environmental or metabolic stresses. The importance of sphingolipids is recognized by the fact that sphingolipid signaling is implicated in the pathobiology of cancer and other human diseases such as diabetes, heart disease, microbial infections, neurological and immune dysfunctions [7],[8],[9],[10],[11].
–
Sphingolipids metabolism and their route of synthesis are highly conserved from yeast to mammalian cells. Studies using the budding yeast Saccharomyces cerevisiae have served in many ways to foster our understanding of sphingolipid dynamics and their role in the regulation of cell cycle, cell integrity, endocytosis, cytoskeleton dynamics and protein turnover [4],[5]. Additionally, sphingolipids have been implicated in the regulation of stress responses and longevity. For instance, yeast mutants lacking Ydc1p (dihydroceramidase) are characterized by increased chronological lifespan (CLS) whereas the overexpression of YDC1 triggers mitochondria and vacuolar fragmentation, apoptosis and accelerated aging in yeast [12]. Genes involved in sphingolipid metabolism (LAG1, YPC1, YSR3, IPT1, and LCB5) show variable expression in senescent and apoptotic cells [13]. More recently, it was shown that the downregulation of sphingolipid synthesis increases yeast CLS in part due to a reduction in long-chain bases (LCBs) mediated activation of Sch9p, the yeast homologue of mammalian ribosomal S6K protein kinase [14]. Furthermore, ceramide synthase (Lag1p) and LCB kinase (Lcb4p) activities decrease upon entry into the stationary phase, leading to a large increase in the levels of LCBs [15].
–
We have previously demonstrated that Isc1p, the yeast orthologue of mammalian neutral sphingomyelinase-2 (nSMase2) responsible for the hydrolysis of complex inositol phosphosphingolipids to produce ceramide, is implicated in oxidative stress resistance and CLS in yeast. Isc1p-deficient cells display shortened CLS, oxidative stress sensitivity and impaired redox and iron homeostasis [16]. Analogous to the role of ceramide and ceramide-activated protein phosphatases in regulating mammalian cell apoptosis, Isc1p acts upstream of Sit4p, the catalytic subunit of protein phosphatase related to type 2A protein phosphatases (PP2A) in yeast. Indeed, SIT4 deletion restores mitochondrial function of isc1Δ cells, increasing oxidative stress resistance and extending CLS [17]. The activation of the HOG (High Osmolarity Glycerol) pathway is also deleterious for isc1Δ cells since ceramide signaling increases the phosphorylation of the Hog1p mitogen activated protein kinase (MAPK) and the disruption of HOG1 attenuates the phenotypes of Isc1p-deficient cells [18].
–
Recent studies also link ceramide to other important signaling pathways involved in the regulation of cell growth and survival, namely the TOR (Target of Rapamycin) pathway. This pathway is highly conserved among organisms, ranging from flies, nematodes, protozoa alongside with mammals [19],[20],[21],[22],[23],[24]. In S. cerevisiae, the TOR pathway is controlled by two Ser/Thr protein kinases, Tor1p and Tor2p, which assemble into two protein complexes with distinct subunit composition and regulatory roles [25],[26],[27]. The rapamycin-sensitive TOR complex 1 (TORC1) contains either Tor1p or Tor2p and is mostly associated with the regulation of cell growth (nutrient sensing), autophagy, ribosomal and protein turnover and cell proliferation [27],[28]. The TOR complex 2 (TORC2) contains Tor2p, but not Tor1p, mediates the proper maintenance of the cell cytoskeleton [29] and regulates ceramide biosynthesis by an Ypk2p-dependent mechanism [30]. Furthermore, TORC2 indirectly modulates Isc1p turnover through the phosphorylation and activation of Slm1p and Slm2p [31].
–
The TORC1 pathway has also been linked to mitochondrial function and yeast CLS [32],[33]. In fact, the deletion of TOR1 or pharmacological inhibition of TORC1 with rapamycin extends CLS in yeast and other organisms [32],[34],[35]. TORC1 is active during early stages of growth and represses the induction of stress responses and entry into the stationary phase, in part by inhibiting the Rim15p protein kinase and consequently the translocation of Msn2p/4p and Gis1p transcription factors into the nucleus [36],[37]. Reducing TORC1 signaling at early stages of growth extends CLS by an intrinsic mechanism involving enhanced mitochondrial membrane potential and superoxide production. This in turn induces an adaptive response that contributes to decrease ROS production in the stationary phase and promotes longevity in yeast [33]. Moreover, reduced TORC1 signaling derepresses Rim15p and triggers the expression of genes regulated by the mitochondrial signaling pathway known as the retrograde response [38],[39],[40] as well as stress-related genes under the control of Msn2p/Msn4p [37],[41].
–
Some authors have identified downstream targets of TORC1 involved in the regulation of stress response and aging, namely the AGC protein kinase Sch9p and the Sit4p protein phosphatase. Apart from sensing nutrient and stress signals from TORC1, both proteins also regulate CLS by integrating sphingolipid signaling. In addition to phosphorylation in the C-terminus mediated by TORC1, Sch9p is phosphorylated in a Thr570 residue in the activation loop by Pkh1/2p protein kinases, homologues of mammalian phosphoinositide-dependent protein kinase 1 (PDK1), in response to LCBs [14],[42]. On the other hand, Sit4p is downregulated by TORC1 in a Tip41p/Tap42p manner [43],[44] but is also activated by ceramide and functions downstream of Isc1p [17]. How this complex network of interacting pathways regulates CLS remains poorly characterized.
–
In this study, we show that the activation of the TORC1-Sch9p pathway impairs oxidative stress resistance, mitochondrial function and CLS in isc1Δ cells. Consistent with TORC1 activation, the autophagic flux is decreased in isc1Δ cells. Our results suggest that Isc1p regulate mitochondrial function and yeast CLS through the TORC1-Sch9p but Isc1p and TORC1 also seem to act through independent pathways. The suppression of isc1D phenotypes by TOR1 deletion is not associated with the attenuation of Hog1p hyperphosphorylation. Our data also support a model in which TORC1 downstream effectors Sit4p and Sch9p act as physiological hubs integrating nutrient and ceramide signaling driven by Isc1p.
RESULTS
The deletion of TOR1 or SCH9 suppresses the shortened CLS and oxidative stress sensitivity of isc1Δ cells
To assess changes in TORC1 signaling associated with Isc1p deficiency, we have monitored TORC1 activity in vivo by assessing TORC1-dependent Sch9p phosphorylation at the C-terminus [45].
–
The results show that Sch9p is hyperphosphorylated in isc1Δ mutants when compared to parental cells, indicative of increased TORC1 activity (Fig. 1A-B). Apart from TORC1, Sch9p can also be phosphorylated by Pkh1/2p in response to LCBs. Since basal levels of phytosphingosine (PHS) are increased in isc1Δ cells during aging [17], we also assessed the Pkh1/2p-dependent phosphorylation of Sch9p on the Thr570 residue. The results show that the levels of Sch9p-phospho-Thr570 were similar in parental and isc1Δ cells (Supplementary Fig. S1).
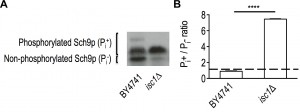 | FIGURE 1: Isc1p-deficient cells exhibit enhanced TORC1 activity in vivo. The TORC1-dependent C-terminal phosphorylation of Sch9p was used to assess TORC1 activity in vivo. (A) S. cerevisiae BY4741 and isc1Δ cells transformed with pJU676 (expressing SCH9-5HA) were grown in SC-medium to the exponential phase and NTCB-treated protein extracts were analyzed by immunoblotting using anti-HA antibody, as described in Materials and Methods. A representative blot out of three is shown. (B) Quantification of band intensities was performed by densitometry. The ratio between phosphorylated and unphosphorylated Sch9p (Pi+/Pi-) is shown. Values are mean ± SD of at least three independent experiments. ****p<0.0001. |
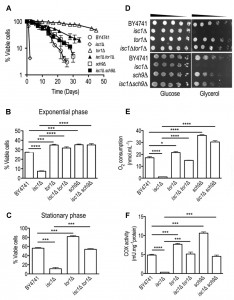
This led us to postulate that TORC1 activation may contribute to isc1Δ phenotypes. To test this hypothesis, we then evaluated if the deletion of TOR1 or SCH9 could abolish the premature aging and hydrogen peroxide sensitivity of isc1Δ cells. In agreement with previous reports [16],[17],[18], isc1Δ cells presented a shortened CLS compared to parental cells (Fig. 2A). Although the acidification of the growth medium due to acetic acid production decreases yeast lifespan [46], it is unlikely that it contributes to the shortened CLS of isc1Δ cells since ISC1 deletion increases acetic acid resistance [47]. As expected, tor1Δ and sch9Δ cells exhibited an increased lifespan (Fig. 2A). The effect of SCH9 deletion was significantly lower to that reported by other groups [48]. This probably results from differences in the growth medium composition, in particular amino acid concentration. Indeed, sch9Δ mutants display increased lifespan when cells are grown in media supplemented with a 3.5-fold excess of amino acids but present shortened lifespan than parental cells when grown in media with 0.5-fold amino acid content [49]. Importantly, the deletion of TOR1 or SCH9 in isc1Δ cells significantly extended the CLS of this mutant but the isc1Δtor1Δ and isc1Δsch9Δ double mutants exhibited a shorter CLS compared to that of tor1Δ and sch9Δ cells, respectively (Fig. 2A).
FIGURE 2: The deletion of TOR1 and SCH9 abolishes the shortened CLS, oxidative stress sensitivity and mitochondrial dysfunctions displayed by isc1Δ cells. (A) S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ, sch9Δ and isc1Δsch9Δ cells were grown in SC-medium and kept in the medium at 26oC. The viability was determined by standard dilution plate counts and expressed as the percentage of the colony-forming units at time T in relation to T0. Values are mean ± SD of at least three independent experiments. (B, C) Yeast cells were grown in SC-medium to the exponential phase (B) or stationary phase (48 hours after exponential phase) (C) and exposed to 1.5 mM H2O2 for 60 min (B) or 300 mM H2O2 for 30 min (C). Cell viability was determined by standard dilution plate counts and expressed as the percentage of the colony-forming units (treated cells versus untreated cells). Values are mean ± SD of at least three independent experiments. ****p<0.0001. ***p<0.001. (D) S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ, sch9Δ and isc1Δsch9Δ cells were grown in SC-medium to the exponential phase and then diluted to OD600=0.1. Fivefold dilution series were spotted on YPD (glucose) or YPG (glycerol) medium and cells were grown at 26oC for 4 days. (E) Oxygen consumption rate was measured in cells grown to PDS phase, as described in Materials and Methods. Values are mean ± SD of at least three independent experiments. ****p<0.0001; *p<0.05. (F) Cytochrome c oxidase (COX) activity was determined in cells grown to the PDS phase. Cells were lysed and enzymatic activity was measured as described in Materials and Methods. Values are mean ± SD of at least three independent experiments. ****p<0.0001; ***p<0.001.
To assess oxidative stress resistance, cells were grown to the exponential or early stationary phase and treated with H2O2. Consistent with published data [50], tor1Δ and sch9Δ cells were more resistant to oxidative stress than parental cells (Fig. 2B-C). The deletion of TOR1 or SCH9 suppressed the hydrogen peroxide sensitivity of isc1Δ cells (Fig. 2B-C). Similar results were obtained by the pharmacological inhibition of TORC1 using rapamycin (Supplementary Fig. S2). These results implicate TORC1-Sch9p activation in the premature aging and oxidative stress sensitivity of Isc1p-deficient cells.
–
Reduced TORC1 signaling enhances mitochondrial coupled respiration in isc1Δ cells
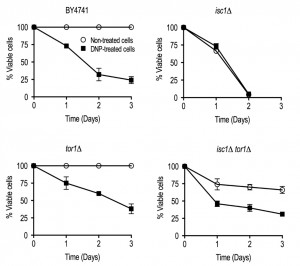
It was previously shown that tor1Δ cells have an extended CLS in part associated with improved and better coupled mitochondrial respiration at early stages of growth, which ultimately preconditions yeast to better survive on the stationary phase [33]. Therefore, we evaluated if the deletion of TOR1 and SCH9 could improve mitochondrial fitness of isc1Δ cells, eventually contributing to CLS extension observed in isc1Δtor1Δ and isc1Δsch9Δ double mutants. To address this hypothesis, we analyzed cell growth in medium containing glycerol, a non-fermentable carbon source, as well as oxygen consumption and cytochrome c oxidase (COX) activity. It was observed that the growth defect of isc1Δ cells on glycerol medium was suppressed upon the deletion of TOR1 or SCH9 (Fig. 2D). As expected, the deletion of ISC1 almost completely abolished oxygen consumption and COX activity in the post-diauxic shift (PDS; respiratory) phase (Fig. 2E-F). In tor1Δ and sch9Δ cells, both oxygen consumption and COX activity were increased when compared to parental cells. Notably, TOR1 and SCH9 disruption suppressed the defects observed in isc1Δ cells: both oxygen consumption and COX activity increased in isc1Δtor1Δ and isc1Δsch9Δ cells, although to levels lower than those observed in tor1Δ and sch9Δ cells, respectively (Fig. 2E-F). To assess if enhanced mitochondrial respiration coupling contributes to the lifespan extension of isc1Δ cells imposed by reduced TORC1 signaling, CLS was analyzed in cells treated with 2,4-dinitrophenol (DNP), which decreases mitochondrial membrane potential and uncouples electron transport from ATP synthesis. Untreated Isc1p-deficient cells exhibited a shortened CLS that was not affected by exposure to DNP (Fig. 3), providing strong evidence that mitochondrial dysfunction largely contributes to isc1Δ phenotypes. In tor1Δ mutants, DNP significantly affected cell viability (e.g., it decreased 40% in DNP-treated vs control cells aged for 2 days). However, the detrimental effect of DNP in tor1Δ mutants was lower to the observed in parental cells (in this strain, the viability of cells treated with DNP and aged for 2 days decreased 68%). This is consistent with published data showing that tor1Δ cells present better coupled respiration when compared to parental cells [33], and therefore are able to mount a more effective adaptive response to counteract DNP effects. The deletion of TOR1 in isc1Δ cells extended CLS (in cells untreated with DNP) and this effect appears to be correlated with an increased coupled respiration imparted by reduced TORC1 signaling since DNP treatment decreased cell viability in such conditions. The isc1Δtor1Δ double mutant presented an intermediate phenotype between parental and tor1Δ cells since, at day 2 of aging, parental, tor1Δ and isc1Δtor1Δ cells presented 32%, 60% and 40% cell viability, respectively. Taking together, we claim that improved mitochondrial fitness (increased coupled respiration) promoted by the deletion of TOR1 per se extends CLS in isc1Δ cells.
FIGURE 3: Increased mitochondrial coupled respiration imparted by reduced TORC1 signaling extends lifespan in isc1Δ cells. S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ cells were grown in SC-medium to the PDS phase, treated with 10 µM 2,4-dinitrophenol (DNP; ■) or vehicle (DMSO; ○) and kept in the medium at 26oC. The viability was determined by standard dilution plate counts and expressed as the percentage of the colony-forming units at time T in relation to T0. Values are mean ± SD of at least three independent experiments.
Hyperpolarization and fragmentation of the mitochondrial network in isc1Δ cells are suppressed by TOR1 or SCH9 deletion
To get further insights into alterations in mitochondrial function operating on isc1Δ cells, we assessed the mitochondrial membrane potential (Δψm), a parameter that has been used to monitor changes on bioenergetics and functions as a key indicator of cell health or injury [51]. For this purpose, yeast cells were labeled with a mitochondria-specific voltage-dependent dye, 3,3-dihexyloxacarbocyanine iodide [DiOC6(3)], which aggregates and preferentially accumulates into functional mitochondria, and analyzed by flow cytometry (Fig. 4A). At the PDS phase, cells lacking Isc1p displayed an enhanced Δψm when compared to parental cells, which is consistent with mitochondrial hyperpolarization. In contrast, tor1Δ and sch9Δ cells had a slightly lower Δψm, which has been associated with mild mitochondrial uncoupling [33]. Both TOR1 and SCH9 disruption in isc1Δ cells reversed the mitochondrial hyperpolarization and decreased the Δψm to values similar to those observed in the respective single mutants. This probably increases cell survival in isc1Δtor1Δ and isc1Δsch9Δ cells, since isc1Δ cells die by caspase-dependent apoptosis upon oxidative stress and during cell aging [16] and mitochondrial hyperpolarization has been associated with the activation of a mitochondrial dependent apoptotic pathway, which initially involves a transient hyperpolarization followed by depolarization of the mitochondrial membrane and release of cytochrome c from the mitochondria into the cytosol [52].
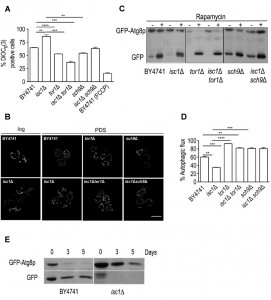 | FIGURE 4: The mitochondrial membrane hyperpolarization and decreased autophagic flux contribute to mitochondrial dysfunction and impairment of mitochondrial dynamics in isc1Δ cells. (A) S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ, sch9Δ and isc1Δsch9Δ cells were grown in SC-medium to the PDS phase, stained with the potential-sensitive dye 3,3-dihexyloxacarbocyanine iodide [DiOC6(3)] for 30 min and analyzed by flow cytometry. Treatment of the parental strain (BY4741) with FCCP (carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone) was used as a positive control (depolarizing event). Values are mean ± SD of at least three independent experiments. ****p<0.0001; ***p<0.001; **p<0.01. (B) Yeast cells transformed with pYX222-mtDsRed were grown to the exponential and PDS phases and analyzed by fluorescence microscopy, as described in Materials and Methods. Live cells were visualized by fluorescence microscopy. A representative experiment out of three is shown. Scale bar: 5 µm. (C) S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ, sch9Δ and isc1Δsch9Δ cells carrying pRS416 GFP-ATG8 were grown to the exponential phase in SC-medium and treated with either rapamycin (200 ng/mL) or DMSO (vehicle) for 3 hours. Proteins were analyzed by immunoblotting, using anti-GFP antibody. (D) The autophagic flux was calculated by the ratio between the free GFP signal and the sum of free GFP and GFP-Atg8p signals. Values are mean ± SD of at least three independent experiments ****p<0.0001; ***p<0.001; **p<0.01. (E) S. cerevisiae BY4741 and isc1Δ cells carrying pRS416 GFP-ATG8 were grown to PDS phase, washed twice with water and then maintained in water. Proteins were analyzed by immunoblotting, using anti-GFP antibody. |
The mitochondrial membrane potential plays a key role in the regulation of mitochondrial morphology and alterations on this parameter were demonstrated to impact on mitochondrial dynamics [53],[54],[55],[56]. To assess changes in mitochondrial network dynamics, yeast cells expressing a mitochondria-targeted DsRed protein were analyzed by fluorescence microscopy. At the exponential phase, the mitochondrial network was not yet fully developed and no significant differences were observed between the parental and isc1Δ cells (Fig. 4B). This was expected since cells are undergoing fermentative growth at this phase. However, in the PDS phase, loss of Isc1p led to the formation of a typical punctuate pattern contrasting with the tubular and well-organized network observed in healthy parental cells (Fig. 4B). This structural alteration has been associated with mitochondrial fragmentation and observed in cells undergoing apoptotic cell death. The normal tubular mitochondrial network was restored upon disruption of TOR1 or SCH9 in isc1Δ cells (Fig. 4B), suggesting that TORC1 and its downstream target Sch9p are also implicated in the regulation of mitochondrial dynamics by promoting network fragmentation.
–
Previous studies have demonstrated that autophagy has an important role in maintaining proper mitochondrial function and dynamics since autophagy-defective mutants present severe mitochondria dysfunctions [57],[58]. In particular, the regulation of the mitochondrial membrane potential appears to be crucial to regulate autophagic flux. In addition, accumulating evidence show that defects in autophagy deregulate mitochondrial dynamics [59]. Since isc1Δ cells present similar phenotypic features and TORC1, a negative regulator of autophagy, is activated in this mutant strain, we evaluated if these cells present autophagy defects. For this purpose, we have monitored the processing of GFP-Atg8p. Once autophagy is induced, GFP-Atg8p is recruited to drive autophagosome biogenesis and then delivered to the vacuole inside the autophagic body. Whereas Atg8p is degraded by resident vacuolar hydrolases, the GFP moiety is relatively resistant to proteolysis. Therefore, the appearance of free GFP signal is indicative of autophagy induction. To induce autophagy, yeast cells were treated with rapamycin. Under these conditions, the autophagic flux was significantly lower in Isc1p-deficient cells (35%) when compared to the parental strain (60%) (Fig. 4C-D). Notably, a slower migrating band (above free GFP) was observed in isc1Δ cells, both under basal conditions and upon rapamycin treatment (see Supplementary Fig. S3 for a longer exposure time of the Western blot). It probably results from an incomplete or aberrant processing of GFP-Atg8p, possibly due to vacuolar dysfunction (defective Pep4p-dependent proteolytic activity) [47] or alterations in vacuolar morphology upon deletion of ISC1 [60]. In isc1Δtor1Δ and isc1Δsch9Δ double mutants, the autophagic flux increased to values close to those observed for the respective tor1Δ and sch9Δ single mutants (Fig. 4C-D). It should be noted that rapamycin is predicted to have still some effect on tor1Δ cells because there is yet some functional TORC1 signaling. In fact, TORC1 complex is sensitive to rapamycin due to binding of FKBP-rapamycin complex to subunits of TORC1. Thus, TORC1 is functional (although with reduced activity) in tor1Δ cells since it may also contain Tor2p (which may replace Tor1p) in its composition. These results implicate TORC1 and its downstream effector, Sch9p, in the deregulation of autophagy, possibly contributing to mitochondrial network fragmentation and impairment of oxidative stress resistance and CLS in isc1Δ cells.
–
To provide further evidence that autophagy is impaired in isc1Δ cells, we have also monitored the autophagic flux during chronological aging. The rate of viability loss during aging of this mutant is very high when cells are grown in SC-medium (Fig. 2A). Thus, to avoid unspecific changes that may occur due to cell death, we assessed autophagy under conditions of calorie restriction (cells grown to PDS phase, washed and maintained in water overtime). We have previously shown that calorie restriction increases CLS in both isc1Δ and parental cells, but isc1Δ mutants still exhibit a premature aging phenotype [17]. The results show that the autophagic flux increased in parental cells aged for 3-5 days, but it was significantly compromised in isc1Δ cells (Fig. 4E). Overall, the data suggests that autophagy is impaired in this mutant strain.
–
TOR1 and SCH9 deletion in isc1Δ cells decrease ROS levels, catalase A deficiency and apoptotic cell death
Apoptosis and aging has been extensively associated with enhanced ROS production [61],[62],[63],[64]. Thus, the improvement of mitochondrial function and/or antioxidant defenses may decrease mitochondrial ROS production or increase its detoxification, leading to CLS extension. To test this hypothesis, ROS levels were measured by flow cytometry using early stationary phase cells stained with dihydroethidium (DHE), a molecular probe sensitive to superoxide radicals. The results show that ROS levels were low in parental, tor1Δ and sch9Δ cells but approximately 50% of isc1Δ cells were DHE-positive (Fig. 5A). In isc1Δtor1Δ and isc1Δsch9Δ mutants, ROS levels were higher than in parental cells but significantly lower when compared to Isc1p-deficient cells (by approximately one-half), suggesting that TOR1 and SCH9 deletion increase survival in isc1Δ cells by decreasing ROS generation.
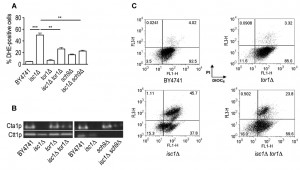 | FIGURE 5: The deletion of TOR1 or SCH9 decreases ROS production, attenuates catalase A deficiency and diminishes apoptotic cell death in isc1Δ cells. (A) S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ, sch9Δ and isc1Δsch9Δ cells were grown to early stationary phase (day 1 in the CLS assay), stained with the dihydroethidium (DHE) for 10 min and analyzed by flow cytometry. Values are mean ± SD of at least three independent experiments. ***p<0.001; **p<0.01. (B) Yeast cells were grown to the PDS phase and catalase activity was detected in situ after non-denaturing polyacrylamide gel electrophoresis, using the H2O2/peroxidase system, as described in Materials and Methods. A representative experiment out of three is shown. (C) BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ cells were double stained with DiOC6(3) and PI (propidium iodide) and analyzed by flow cytometry, as described in Materials and Methods. Representative histograms are shown. |
Increased ROS levels have been associated with homeostatic imbalance partially dictated by impaired cellular antioxidant defences. Hence, we hypothesized that the improvement of the mitochondrial function and dynamics in isc1Δtor1Δ and isc1Δsch9Δ cells may contribute to upregulate antioxidant defence mechanisms and decrease ROS levels during the aging process. It was previously shown that isc1Δ cells fail to induce CTA1 gene expression in the PDS phase [65] and display a low activity of Cta1p [17], the catalase A form present in mitochondria and peroxisomes. Moreover, CTA1 overexpression partially suppresses isc1Δ phenotypes [17]. Our results show that Cta1p activity was partially restored in isc1Δtor1Δ and isc1Δsch9Δ cells (Fig. 5B). In sch9Δ and isc1Δsch9Δ cells, Ctt1p (cytosolic catalase) activity was not detected, which is consistent with the fact that the Sch9p kinase is directly or indirectly involved in the transcriptional control of CTT1 expression in yeast [66]. The analysis of superoxide dismutase activity did not reveal changes in Sod1p (cytosolic form) or Sod2p (mitochondrial form) activity upon the deletion of TOR1 or SCH9 in isc1Δ cells (data not shown).
–
To evaluate if the decrease in ROS production and enhancement of antioxidant defenses (Cta1p) contribute to decrease apoptotic cell death in isc1Δtor1Δ mutants, early stationary phase cells were labeled with DiOC6(3)/propidium iodide (PI). The analysis of yeast cells by flow cytometry allows the definition of four distinct populations: healthy cells (DiOC6(3)-positive/PI-negative), early apoptosis (DiOC6(3)-negative/PI-negative), late apoptosis (DiOC6(3)-positive/PI-positive) and necrosis (DiOC6(3)-negative/PI-positive). The results (Fig. 5C) clearly show that the large majority of parental and tor1Δ cells remained healthy whereas 62% of the isc1Δ cell population was already undergoing early (15%) or late (47%) apoptosis. In the isc1Δtor1Δ double mutant, however, a significant decrease of apoptotic markers was observed, particularly at late stages where a reduction of approximately 50% (from 47% to 24%) was detected.
–
SCH9 deletion but not reduced TORC1 signaling in isc1Δ cells attenuates Hog1p activation
The hyperactivation of the HOG signaling pathway was previously implicated in the premature aging and mitochondrial dysfunction exhibited by Isc1p-deficient cells [18].
–
Thus, we hypothesized that the suppression of isc1Δ phenotypes by TOR1 and SCH9 deletion could be associated with the modulation of the HOG pathway. To address this question, Hog1p phosphorylation was monitored by Western blotting using an anti-phospho-p38 antibody that recognizes dually phosphorylated Hog1p, the active form of this kinase [67],[68]. As illustrated in Fig. 6A, Hog1p phosphorylation was increased in isc1Δ cells when compared to parental cells. In contrast, phosphorylated Hog1p could not be detected in tor1Δ cells. Notably, Hog1p phosphorylation levels in isc1Δtor1Δ cells were even higher to those detected in isc1Δ cells, suggesting a functional interplay between the HOG and TORC1 signaling pathways in isc1Δ cells.
FIGURE 6: The deletion of TOR1 increases Hog1p phosphorylation without affecting its cytosolic localization whereas SCH9 disruption diminishes Hog1p phosphorylation in isc1Δ cells. (A) Hog1p activation was monitored in BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ cells by immunoblotting, using anti-phospho-p38 antibody (top panel) that detects the phosphorylated form of Hog1p, or anti-Pgk1p (loading control) as primary antibodies. A representative blot out of three is shown. (B) S. cerevisiae BY4741, isc1Δ, tor1Δ and isc1Δtor1Δ cells expressing the consensus Rlm1p binding sequences fused to a LacZ reporter (2xRLM1-LacZ), were grown to the exponential phase and the β-galactosidase activity was measured as described in Materials and Methods. Values are mean ± SD of at least three independent experiments. ****p<0.0001; ***p<0.001; *p<0.05. (C) Hog1p activation was monitored in BY4741, isc1Δ, sit4Δ and isc1Δsit4Δ cells by immunoblotting, as described in A. A representative blot out of three is shown. (D) Hog1p activation was monitored in BY4741, isc1Δ, sch9Δ and isc1Δsch9Δ cells by immunoblotting, as described in A. BY4741 and sch9Δ cells were grown to the exponential phase and treated with either 10 μM C2-ceramide or DMSO (vehicle) for 1 h. A representative blot out of three is shown.
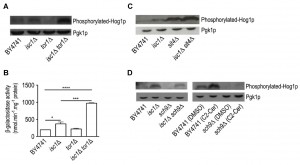
It was previously shown that Hog1p activation leads to its import into the nucleus where it phosphorylates the Msn2p/Msn4p, Hot1p, Sko1p and Smp1p transcription factors to promote adaptation to stress conditions [69],[70],[71],[72],[73],[74],[75],[76]. Thus, we have also monitored Hog1p cell localization by fluorescence microscopy in cells expressing Hog1p-GFP (Supplementary Fig. S4). Similarly to parental cells, Hog1p was present in the cytoplasm of isc1Δ, tor1Δ and isc1Δtor1Δ mutants, implying that TORC1 may not regulate Hog1p localization. We have also evaluated the activation of the Cell Wall Integrity (CWI) pathway, since we have previously reported a Hog1p-dependent activation of Slt2p, a MAPK of the CWI pathway in isc1Δ cells [18]. For this purpose, we monitored the activation of Rlm1p, a transcription factor regulated by Slt2p, by measuring β-galactosidase activity in cells expressing a LacZ reporter under the control of Rlm1p promoter. Consistent with the hyperactivation of Hog1p and induction of the Rlm1p-driven LacZ reporter, β-galactosidase activity was increased by 1.7- and 4.3-fold in isc1Δ and isc1Δtor1Δ cells, respectively (Fig. 6B).
–
It was previously shown that the protein phosphatase Sit4p is negatively regulated by TORC1 signaling [44] but activated by ceramide [77]. Moreover, the deletion of SIT4 suppresses isc1Δ phenotypes [17]. Notably, Hog1p phosphorylation increased in sit4D cells and it was exacerbated in isc1Δsit4Δ cells (Fig. 6C), as observed in isc1Δtor1Δ cells (Fig. 6A). This suggests that the phosphorylation of Hog1p is regulated by a Sit4p-dependent mechanism.
–
Remarkably, the deletion of SCH9 decreased Hog1p phosphorylation in isc1Δ cells (Fig. 6D). It was previously reported that ceramide signaling increases Hog1p phosphorylation [18]. Thus, we tested if Sch9p regulates the HOG pathway in response to ceramide. As previously reported, C2-ceramide treatment increased Hog1p phosphorylation in parental cells (Fig. 6D). In contrast, the deletion of SCH9 completely abolished Hog1p activation upon treatment with ceramide (Fig. 6D). These results suggest that Sch9p is acting upstream of Hog1p in response to ceramide signaling.
–
Overall, the results show that the deletion of TOR1 or SIT4 does not suppress isc1Δ phenotypes through the attenuation of Hog1p hyperactivation and Sch9p appears to regulate the activation of the HOG pathway in response to ceramide in Isc1p-deficient cells.
DISCUSSION
The TORC1 pathway is a well-established nutrient response pathway that modulates aging and age-related diseases [23]. Here we provided evidence that TORC1 signaling is deregulated in cells lacking Isc1p, the yeast orthologue of mammalian neutral sphingomyelinase. Isc1p-deficient cells exhibit increased TORC1 activity, which is detrimental for this mutant. In fact, the deletion of TOR1 alleviates the premature aging, oxidative stress sensitivity and mitochondrial dysfunctions of isc1Δ cells. However, isc1Δtor1Δ cells exhibit lower resistance to oxidative stress, shortened CLS, slightly impaired mitochondrial function, and higher levels of ROS and apoptotic cell death markers compared to the tor1Δ mutant strain, suggesting that TORC1-independent mechanisms also contributes to isc1Δ phenotypes. In agreement, the overexpression of ISC1 does not suppress the rapamycin hypersensitivity of tor1Δ mutants, suggesting that Isc1p is not acting downstream of TORC1 (Supplementary Fig. S5).
–
Several lines of evidence suggest an intricate interplay between sphingolipid metabolism and TOR signaling. Both TORC1 and TORC2 control the biosynthesis of sphingolipids through regulation of Orm1p and Orm2p, two evolutionarily conserved integral membrane proteins of the endoplasmic reticulum [78],[79],[80]. However, TORC1 and TORC2 signaling seems to function independently in the regulation of sphingolipid metabolism. TORC1 negatively controls the synthesis of complex sphingolipids from ceramide via inhibition of Orm1/2p phosphorylation in a process mediated by Npr1p [80]. TORC2 stimulates the de novo biosynthesis of sphingolipids via activation of Ypk1p, which then phosphorylates and inactivates Orm1/2p [81]. Since Orm1/2p bind to and inhibit serine palmitoyl-coenzyme A transferase (SPT) [82], which catalyzes the first step in sphingolipid biosynthesis [83], its inactivation by a Ypk1p-dependent phosphorylation alleviates SPT inhibition and increases sphingolipid production. In addition, TORC2 stimulates ceramide biosynthesis by an Ypk2p-dependent mechanism [30] and inhibits Isc1p activity via modulation of Slm1/2p [31],[84].
–
Our data suggests a genetic interaction between ISC1 and TOR1. A recent report suggests a link between ceramide generated by mammalian acid sphingomyelinase under amino acid deprivation conditions and mTOR inactivation by a PP1/PP2A-dependent mechanism [85]. Notably, yeast Isc1p appears to occupy a central position and may possibly act as a metabolic hub (together with ceramide synthase) in the TOR pathway to regulate ceramide dynamics, since Isc1p is functionally linked with both TORC1 and TORC2 branches. In particular, the modulation of complex sphingolipids turnover by coupling TORC1-regulated biosynthesis with Isc1p-driven hydrolysis may be important to control ceramide flux in yeast.
–
The molecular mechanism by which TORC1 is activated in isc1Δ cells remains to be elucidated. Nevertheless, the activation of TORC1 in Isc1p-deficient cells may be part of a homeostatic response that aims to decrease the synthesis of complex sphingolipids, since isc1Δ cells accumulate inositolphosphorylceramide (IPC) and mannosyldiinositolphosphorylceramide (M(IP)2C) [31],[86] and TORC1 negatively controls their biosynthesis [80]. However, TORC1 activation impairs mitochondrial function and oxidative stress resistance in isc1Δ cells, and TOR1 deletion suppresses isc1Δ phenotypes. Such features are in agreement with previous studies showing that TOR1 deletion regulates yeast CLS by a cell-intrinsic mechanism [32],[33]. Reduced TORC1 signaling increases mitochondrial coupling during active growth in yeast cells, eliciting an adaptive response that preconditions yeast cells to better survive in the stationary phase and promotes longevity [33]. Furthermore, TOR1 disruption leads to an increased translation of mtDNA-encoded subunits of the oxidative phosphorylation system [32], which is consistent with the higher oxygen consumption and COX activity observed in this study. In addition, it improves oxidative stress resistance in the stationary phase [50].
–
In the present study, we demonstrate that TORC1-driven Sch9p C-terminal phosphorylation is increased in isc1Δ cells and our results are consistent with TORC1 acting through Sch9p-associated mechanisms since the disruption of both TOR1 and SCH9 abolishes isc1Δ phenotypes. Importantly, Isc1p-deficient cells presented reduced autophagic flux, both upon treatment with rapamycin and during cell aging, which is consistent with the activation of the TORC1-Sch9p pathway. In agreement, TOR1 and SCH9 disruption reestablished the functional integrity of autophagy in isc1Δ cells and this is correlated with the restoration of mitochondrial function in the double mutants, ultimately abolishing the H2O2 hypersensitivity and premature aging exhibited by this mutant strain.
–
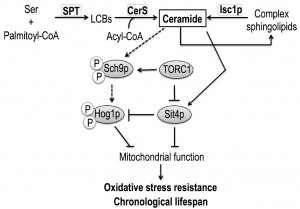
It was previously demonstrated that the accumulation of very long chain ceramide species (dihydro-C26-ceramide and phyto-C26-ceramide species) and ceramide-activation of the protein phosphatase Sit4p are also implicated in isc1Δ phenotypes [17]. Since TORC1 negatively regulates Sit4p by promoting its association with the inhibitor Tap42p [43] and both TOR1 and SIT4 deletions suppress isc1Δ phenotypes, we propose that Sit4p may act in parallel to TORC1 signaling, by directly sensing ceramide signals (Fig. 7).
FIGURE 7: Sit4p and Sch9p integrate nutrient and stress signaling from TORC1 with ceramide signals to regulate mitochondrial function and CLS in yeast. Cells lacking Isc1p present increased TORC1-Sch9p activity, ultimately leading to mitochondrial dysfunction, decreased oxidative stress resistance and shortened CLS. Apart from sensing signals from TORC1, the ceramide activated protein phosphatase Sit4p and the AGC protein kinase Sch9p also integrate ceramide signaling derived from Isc1p to regulate mitochondrial function, oxidative stress resistance and chronological lifespan in yeast.
Several pieces of evidence suggest a functional link between sphingolipids, TOR signaling and the HOG pathway. The constitutive activation of Hog1p is deleterious for isc1Δ cells since the deletion of HOG1 attenuates the shortened CLS, hydrogen peroxide sensitivity and mitochondrial dysfunctions of this mutant strain [18]. The HOG pathway is activated upon treatment of yeast cells with myriocin, an inhibitor of the de novo sphingolipid biosynthesis, and in cells with impaired synthesis of IPC [87]. In addition, reduced TOR signaling lowers the basal activity of Hog1p in Candida albicans through the Hog1p tyrosine phosphatases Ptp2 and Ptp3 [88] and our results show a decrease of Hog1p basal phosphorylation in the tor1Δ mutant. In this study, we demonstrate that TOR1 deletion does not suppress Hog1p hyperactivation or alter Hog1p localization in isc1Δ cells. However, our results support a more intricate interplay between the two signaling pathways in isc1Δ cells. Indeed, Hog1p phosphorylation was even increased in isc1Δtor1Δ cells in comparison to isc1Δ cells and remarkably a similar feature was also observed for both sit4Δ and isc1Δsit4Δ cells, which present extended lifespan when compared to isc1Δ or even to parental cells [17]. We propose that the deletion of TOR1 or SIT4 either suppresses putative deleterious effects of Hog1p hyperactivation on mitochondrial function and CLS or modulates the regulatory role of Hog1p to determine cell fate in Isc1p-deficient cells. In fact, the activation of yeast Hog1p or its mammalian orthologue, p38 MAPK, has been implicated in both cell survival and cell death. For example, HOG1 deletion decreases osmotic and oxidative stress resistance and shortens CLS in yeast [89],[90]. However, the expression of constitutively active Hog1p mutant kinases or constitutive activation of the HOG pathway is lethal [91],[92],[93], and hyperactivation of Hog1p is detrimental under heat stress conditions [94]. In mammalian cells, p38 MAPK is activated during the onset of senescence [95],[96], its inhibition moderately delays replicative senescence [97] and its activation promotes apoptosis [98],[99].
–
Remarkably, we provide significant evidence that the activation of the HOG pathway is modulated by ceramide signaling by Sch9p-dependent mechanisms, ultimately contributing to isc1Δ phenotypes. Indeed, C2-ceramide-induced Hog1p phosphorylation [18] was suppressed by the disruption of SCH9. In addition, Hog1p phosphorylation decreased upon deletion of SCH9 in Isc1p-deficient cells. Overall, the results suggest that both proteins act in the same pathway since SCH9 (this study) and HOG1 [18] deletions suppressed isc1Δ phenotypes. Therefore, our results points out to a model in which Sch9p integrates nutrient and stress signals from TORC1 with ceramide signaling, the latter contributing to the modulation of the Hog1p phosphorylation. Interestingly, this appears to be ceramide-specific since treatment with phytosphingosine had no effect on Hog1p phosphorylation (data not shown). It is known that Sch9p possesses a C2-domain, which is known to bind to several substrates, namely Ca2+, phospholipids, inositol polyphosphates, and intracellular proteins [100]. Whether ceramide acts by binding directly to the C2-domain remains to be established.
–
In summary, our data implicate TORC1-Sch9p activation in the mitochondrial dysfunction, premature aging, oxidative stress sensitivity and impaired autophagy exhibited by isc1Δ cells. However, Isc1p and TORC1 also seem to act through independent pathways, as isc1Δtor1Δ phenotypes are intermediate to those displayed by isc1Δ and tor1Δ cells.
–
The lifespan extension in isc1Δ cells imparted by reduced TORC1 signaling is not associated with the attenuation of Hog1p hyperphosphorylation. Our results suggest that complex signaling interconnections involving TORC1 and its downstream effectors Sch9p and Sit4p govern the redox homeostasis and lifespan of isc1Δ cells and support a model in which both proteins act as physiological core centers integrating nutrient and stress signal from TORC1 and ceramide signaling derived from Isc1p to regulate mitochondrial function and CLS in yeast (Fig. 7).
MATERIALS AND METHODS
Yeast cells and growth conditions
S. cerevisiae BY4741 was the parental strain of all haploid derivatives used in this study (Table 1). Yeast cells were grown aerobically at 26°C in a gyratory shaker (at 140 r.p.m.), with a ratio of flask volume/ medium volume of 5:1. The growth media used were YPD [1% (w/v) yeast extract, 2% (w/v) bactopeptone, 2% (w/v) glucose], YPGlycerol [1% (w/v) yeast extract, 2% (w/v) bactopeptone, 4% (v/v) glycerol] or synthetic complete (SC) drop-out medium containing 2% (w/v) glucose, 0.67% yeast nitrogen base without amino acids and supplemented with appropriate amino acids (80 mg histidine L-1, 400 mg leucine L-1, 80 mg tryptophan L-1 and 80 mg uracil L-1). The tor1Δ cells were obtained by replacing TOR1 with a tor1::KanMX4 deletion cassette amplified by polymerase chain reaction (PCR) using the corresponding strain available from EUROSCARF (Germany) and the following primers: Fw (GA GAATCATTACCGGCGAAA) and Rv (ACGAACACGTTTTGGTG ATG). For ISC1 disruption in tor1Δ and sch9Δ::KanMX4 cells, a deletion fragment containing LEU2 and the flanking regions of ISC1 was amplified by PCR using pRS315 and the following primers: Fw (ATTTGCGCTTTCCGTAAAAAGGGAAAAAAAGCA GATATTTAAGCAAGGATTTTCT) and Rv (TCAGTAATTTTTTTACA TATGCTAAAGAAAATCGATAATACCGCATATCGACCCTCGAGGA G). Cells were transformed by electroporation. The tor1Δ cells were selected in YPD medium containing geneticin (0.4 mg.mL-1) whereas isc1Δtor1Δ and isc1Δsch9Δ cells were selected in minimal medium lacking leucine [0.67% (w/v) yeast nitrogen base without amino acids, 2% (w/v) glucose supplemented with appropriate amino acids (40 mg histidine L-1, 40 mg uracil L-1 and 40 mg methionine L-1)]. The correct insertion of all cassettes was confirmed by PCR.
TABLE 1. Saccharomyces cerevisiae strains used in this study. Harboring plasmids are shown in square brackets. [17]

To evaluate TORC1-dependent C-terminal phosphorylation of Sch9p, BY4741 and isc1∆ cells were transformed by electroporation with pJU676 [45] and selected in minimal medium lacking uracil. For the analysis of mitochondrial morphology, yeast cells were transformed with a plasmid expressing mitochondrial DsRed (pYX222-mtDsRed) and selected in minimal medium lacking histidine. To evaluate autophagic flux, cells were transformed by electroporation with pRS416-GFP-ATG8 [101] and selected in minimal medium lacking uracil. For Hog1p localization assays, yeast cells were transformed with pRS416-HOG1-GFP [102] and selected in minimal medium lacking uracil. To assess the activation of the CWI pathway by Rlm1p reporter transcriptional activity, yeast cells were transformed with pLGD312-2xRLM1-LacZ [103] and selected in minimal medium lacking uracil. For epistatic analysis, BY4741 and tor1Δ strains were transformed with pYES2 and pYES2-ISC1 [16] and selected in minimal medium lacking uracil.
–
Stress resistance and chronological lifespan
For H2O2 resistance assay, cells were grown to the exponential phase (OD600 = 0.6) or early stationary phase (48h after exponential phase) and exposed to 1.5 mM or 300 mM H2O2 (Merck) for 60 and 30 min, respectively. The CLS assay was performed as described [104]. For both assays, cell viability was determined by standard dilution plate counts on YPD medium containing 1.5 % (w/v) agar and expressed as the percentage of the colony-forming units after growth at 26°C for 3 days (time T vs. T0 (when viability was considered 100%) for the CLS assay; treated vs. untreated cells for H2O2 resistance). Values are mean ± SD of at least three independent experiments.
–
Enzymatic activities and oxygen consumption
For enzyme activities, yeast cells were harvested by centrifugation for 5 min at 4,000 r.p.m. (4°C). Cells were then resuspended in 50 mM potassium phosphate buffer (pH 7.0) containing protease inhibitors (Complete, Mini, EDTA-free Protease Cocktail Inhibitor Tablets; Boehringer Mannhein) and phosphatase inhibitors (50 mM sodium fluoride, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate). Total protein extracts were obtained by mechanical disruption through vigorous shaking of the cell suspension in the presence of glass beads for 5 min. Short pulses of 1 min were applied followed by 1 min incubation on ice. Cell debris was removed by centrifugation at 13,000 r.p.m. for 15 min and protein content was determined by the method of Lowry, using bovine serum albumin as a standard. Catalase activity was analyzed in situ, in the presence of 3,3′-diaminobenzidine tetrahydrochloride, using the H2O2/peroxidase system [105]. Cytochrome c oxidase (COX) activity was determined by measuring cytochrome c oxidation [106]. β-galactosidase activity determination was performed as previously reported [18]. Oxygen consumption rate was measured for 3 x 108 cells in PBS buffer (pH 7.4), using an oxygen electrode (Oxygraph, Hansatech). Data was analyzed using the Oxyg32 V2.25 software.
–
Mitochondrial membrane potential, ROS levels and cell death
The mitochondrial membrane potential was measured using the potential-sensitive dye DiOC6(3). Briefly, 2×106 cells were resuspended in sample buffer [10 mM 2-(N-morpholino) ethanesulfonic acid, 0.1 mM MgCl2 and 2% (w/v) glucose, pH 6.0]. DiOC6(3) (Molecular Probes) was added to a final concentration of 1 nM. The cell suspension was then incubated for 30 min at 26°C, collected by centrifugation and washed twice with PBS. Fluorescence was measured on the FL-1 channel of a Becton Dickinson FACS Calibur Analytic Flow cytometer with excitation and emission settings of 488 nm and 515–545 nm, respectively, without compensation. For the quantification of ROS levels, 5×106 cells were resuspended in PBS and the superoxide anion sensitive probe dihydroethidium (DHE, Molecular Probes) was added to a final concentration of 5 μM. Cells were incubated for 10 min at 26°C, pelleted by centrifugation, washed twice with PBS and analyzed by flow cytometry with excitation and emission settings of 488 nm and ≥670 nm (FL-3 channel), without compensation.
–
For the characterization of the cell death process, cells were dually stained with DiOC6(3) (1 nM) and propidium iodide (PI, 2μg mL-1, Molecular Probes) to evaluate mitochondrial membrane polarization and the plasma membrane integrity, respectively. Cells were incubated for 30 min at 26°C and harvested as previously described. After suitable compensation, fluorescence was measured by flow cytometry at different wavelengths: excitation/emission at 488/525 nm for DiOC6(3) (FL-1 channel), and at 536/600 nm for PI (FL-3 channel). Data treatment was performed using the FlowJo software (Tree Star).
–
Western Blot analysis
To evaluate the TORC1-dependent C-terminal phosphorylation of Sch9p, cells transformed with pJU676 (expressing SCH9-5HA) were grown in SC-medium lacking uracil to the exponential phase. NTCB-chemical fragmentation analysis was done as described [45]. Proteins were then analyzed by SDS-PAGE using 10% polyacrylamide gels and blotted onto a nitrocellulose membrane (GE Healthcare, Buckinghamshire, United Kingdom). Immunodetection was performed using rabbit anti-HA (Sigma-Aldrich) at a 1:1,000 dilution as primary antibody, goat anti-rabbit IgG-peroxidase (Sigma-Aldrich) at a 1:5,000 dilution as secondary antibody, and the Lumigen HRP chemiluminescent substrate (GE Healthcare, RPN2109, Buckinghamshire, United Kingdom).
–
To monitor Hog1p phosphorylation, yeast cells were grown to the exponential phase and protein extracts (50 μg) were separated by SDS-PAGE and blotted onto a nitrocellulose membrane (GE Healthcare, Buckinghamshire, United Kingdom). The membrane was incubated with the primary antibodies rabbit anti-phospho-p38 MAPK (Cell Signaling Technology, Denver, USA) at a 1:500 dilution or mouse anti-Pgk1p (Invitrogen, Carlsbad, USA) at a 1:30,000 dilution. Subsequently, the membrane was incubated with the secondary antibodies, anti-rabbit IgG-peroxidase (Sigma-Aldrich) at a 1:5,000 dilution, or anti-mouse IgG-peroxidase (Molecular Probes) at a 1:3,000 dilution. Immunodetection was performed as described above.
–
The evaluation of LCBs mediated Phk1/2-dependent phosphorylation of Sch9p was performed as described [14]. To assess alterations in autophagic flux, cells were grown to the exponential phase in SC-medium and treated with either rapamycin (200 ng mL-1, (Sigma-Aldrich) or DMSO (vehicle, Sigma-Aldrich) for 3 hours. For CLS assay, cells were grown to the PDS phase, washed twice with water and then maintained in water overtime. Total protein extracts (30 μg) were analyzed in similar conditions as previously described. The membrane was incubated with the primary antibodies mouse anti-GFP (Roche, Basel, Switzerland) at a 1:3,000 dilution or mouse anti-Pgk1p (Invitrogen, Carlsbad, USA) at a 1:30,000 dilution. Subsequently, the membrane was incubated with the secondary antibody anti-mouse IgG-peroxidase (Molecular Probes) at a 1:3,000 dilution. Immunodetection was done as described.
–
Fluorescence microscopy
For mitochondrial morphology analysis, cells transformed with pYX222-mtDsRed were grown in SC-medium lacking histidine to the exponential or PDS phase. To assess Hog1p cell localization, cells expressing Hog1p-GFP were grown in SC-medium lacking uracil to the exponential phase. Live cells were observed by fluorescence microscopy (AxioImager Z1, Carl Zeiss). Data image stacks were deconvolved by QMLE algorithm of Huygens Professional v3.0.2p1 (Scientific Volume Imaging B.V.). Maximum intensity projection was used to output final images using ImageJ 1.45v software.
–
Statistical analysis
Data are expressed as mean values ± SD of at least three independent experiments. Values were compared by Student’s t-test. The 0.05 probability level was chosen as the point of statistical significance throughout. Statistical analyses were carried out using GraphPad Prism Software v5.01 (GraphPad Software).
References
- R.C. Dickson, and R.L. Lester, "Metabolism and selected functions of sphingolipids in the yeast Saccharomyces cerevisiae", Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, vol. 1438, pp. 305-321, 1999. http://dx.doi.org/10.1016/s1388-1981(99)00068-2
- G.M. Jenkins, and Y.A. Hannun, "Role for de Novo Sphingoid Base Biosynthesis in the Heat-induced Transient Cell Cycle Arrest of Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 276, pp. 8574-8581, 2001. http://dx.doi.org/10.1074/jbc.M007425200
- S. Spiegel, and S. Milstien, "Sphingosine-1-phosphate: an enigmatic signalling lipid", Nature Reviews Molecular Cell Biology, vol. 4, pp. 397-407, 2003. http://dx.doi.org/10.1038/nrm1103
- R.C. Dickson, "Thematic Review Series: Sphingolipids. New insights into sphingolipid metabolism and function in budding yeast", Journal of Lipid Research, vol. 49, pp. 909-921, 2008. http://dx.doi.org/10.1194/jlr.R800003-JLR200
- Y.A. Hannun, and L.M. Obeid, "Principles of bioactive lipid signalling: lessons from sphingolipids", Nature Reviews Molecular Cell Biology, vol. 9, pp. 139-150, 2008. http://dx.doi.org/10.1038/nrm2329
- G. van Meer, D.R. Voelker, and G.W. Feigenson, "Membrane lipids: where they are and how they behave", Nature Reviews Molecular Cell Biology, vol. 9, pp. 112-124, 2008. http://dx.doi.org/10.1038/nrm2330
- T. Kolter, and K. Sandhoff, "Sphingolipid metabolism diseases", Biochimica et Biophysica Acta (BBA) - Biomembranes, vol. 1758, pp. 2057-2079, 2006. http://dx.doi.org/10.1016/j.bbamem.2006.05.027
- F.B.K. Ozbayraktar, and K.O. Ulgen, "Molecular facets of sphingolipids: Mediators of diseases", Biotechnology Journal, vol. 4, pp. 1028-1041, 2009. http://dx.doi.org/10.1002/biot.200800322
- T. Kolter, "A view on sphingolipids and disease", Chemistry and Physics of Lipids, vol. 164, pp. 590-606, 2011. http://dx.doi.org/10.1016/j.chemphyslip.2011.04.013
- T. Hla, and A. Dannenberg, "Sphingolipid Signaling in Metabolic Disorders", Cell Metabolism, vol. 16, pp. 420-434, 2012. http://dx.doi.org/10.1016/j.cmet.2012.06.017
- M.M. Young, M. Kester, and H. Wang, "Sphingolipids: regulators of crosstalk between apoptosis and autophagy", Journal of Lipid Research, vol. 54, pp. 5-19, 2013. http://dx.doi.org/10.1194/jlr.R031278
- A.M. Aerts, P. Zabrocki, I.E.J.A. François, D. Carmona-Gutierrez, G. Govaert, C. Mao, B. Smets, F. Madeo, J. Winderickx, B.P.A. Cammue, and K. Thevissen, "Ydc1p ceramidase triggers organelle fragmentation, apoptosis and accelerated ageing in yeast", Cellular and Molecular Life Sciences, vol. 65, pp. 1933-1942, 2008. http://dx.doi.org/10.1007/s00018-008-8129-8
- P. LAUN, L. RAMACHANDRAN, S. JAROLIM, E. HERKER, P. LIANG, J. WANG, M. WEINBERGER, D. BURHANS, B. SUTER, and F. MADEO, "A comparison of the aging and apoptotic transcriptome of", FEMS Yeast Research, vol. 5, pp. 1261-1272, 2005. http://dx.doi.org/10.1016/j.femsyr.2005.07.006
- X. Huang, J. Liu, and R.C. Dickson, "Down-Regulating Sphingolipid Synthesis Increases Yeast Lifespan", PLoS Genetics, vol. 8, pp. e1002493, 2012. http://dx.doi.org/10.1371/journal.pgen.1002493
- R.L. Lester, B.R. Withers, M.A. Schultz, and R.C. Dickson, "Iron, glucose and intrinsic factors alter sphingolipid composition as yeast cells enter stationary phase", Biochimica et Biophysica Acta (BBA) - Molecular and Cell Biology of Lipids, vol. 1831, pp. 726-736, 2013. http://dx.doi.org/10.1016/j.bbalip.2012.12.012
- T. Almeida, M. Marques, D. Mojzita, M.A. Amorim, R.D. Silva, B. Almeida, P. Rodrigues, P. Ludovico, S. Hohmann, P. Moradas-Ferreira, M. Côrte-Real, and V. Costa, "Isc1p Plays a Key Role in Hydrogen Peroxide Resistance and Chronological Lifespan through Modulation of Iron Levels and Apoptosis", Molecular Biology of the Cell, vol. 19, pp. 865-876, 2008. http://dx.doi.org/10.1091/mbc.E07-06-0604
- A.D. Barbosa, H. Osório, K.J. Sims, T. Almeida, M. Alves, J. Bielawski, M.A. Amorim, P. Moradas‐Ferreira, Y.A. Hannun, and V. Costa, "Role for Sit4p‐dependent mitochondrial dysfunction in mediating the shortened chronological lifespan and oxidative stress sensitivity of Isc1p‐deficient cells", Molecular Microbiology, vol. 81, pp. 515-527, 2011. http://dx.doi.org/10.1111/j.1365-2958.2011.07714.x
- A.D. Barbosa, J. Graça, V. Mendes, S.R. Chaves, M.A. Amorim, M.V. Mendes, P. Moradas-Ferreira, M. Côrte-Real, and V. Costa, "Activation of the Hog1p kinase in Isc1p-deficient yeast cells is associated with mitochondrial dysfunction, oxidative stress sensitivity and premature aging", Mechanisms of Ageing and Development, vol. 133, pp. 317-330, 2012. http://dx.doi.org/10.1016/j.mad.2012.03.007
- B. Raught, A. Gingras, and N. Sonenberg, "The target of rapamycin (TOR) proteins", Proceedings of the National Academy of Sciences, vol. 98, pp. 7037-7044, 2001. http://dx.doi.org/10.1073/pnas.121145898
- C. De Virgilio, and R. Loewith, "The TOR signalling network from yeast to man", The International Journal of Biochemistry & Cell Biology, vol. 38, pp. 1476-1481, 2006. http://dx.doi.org/10.1016/j.biocel.2006.02.013
- S.G. Dann, and G. Thomas, "The amino acid sensitive TOR pathway from yeast to mammals", FEBS Letters, vol. 580, pp. 2821-2829, 2006. http://dx.doi.org/10.1016/j.febslet.2006.04.068
- M. Laplante, and D. Sabatini, "mTOR Signaling in Growth Control and Disease", Cell, vol. 149, pp. 274-293, 2012. http://dx.doi.org/10.1016/j.cell.2012.03.017
- S.C. Johnson, P.S. Rabinovitch, and M. Kaeberlein, "mTOR is a key modulator of ageing and age-related disease", Nature, vol. 493, pp. 338-345, 2013. http://dx.doi.org/10.1038/nature11861
- M. Markaki, and N. Tavernarakis, "Metabolic Control by Target of Rapamycin and Autophagy during Ageing - A Mini-Review", Gerontology, vol. 59, pp. 340-348, 2013. http://dx.doi.org/10.1159/000348599
- R. Loewith, E. Jacinto, S. Wullschleger, A. Lorberg, J.L. Crespo, D. Bonenfant, W. Oppliger, P. Jenoe, and M.N. Hall, "Two TOR Complexes, Only One of which Is Rapamycin Sensitive, Have Distinct Roles in Cell Growth Control", Molecular Cell, vol. 10, pp. 457-468, 2002. http://dx.doi.org/10.1016/S1097-2765(02)00636-6
- R. Loewith, and M.N. Hall, "Target of Rapamycin (TOR) in Nutrient Signaling and Growth Control", Genetics, vol. 189, pp. 1177-1201, 2011. http://dx.doi.org/10.1534/genetics.111.133363
- J. Kim, and K. Guan, "Amino Acid Signaling in TOR Activation", Annual Review of Biochemistry, vol. 80, pp. 1001-1032, 2011. http://dx.doi.org/10.1146/annurev-biochem-062209-094414
- D.S. Evans, P. Kapahi, W. Hsueh, and L. Kockel, "TOR signaling never gets old: Aging, longevity and TORC1 activity", Ageing Research Reviews, vol. 10, pp. 225-237, 2011. http://dx.doi.org/10.1016/j.arr.2010.04.001
- N. Cybulski, and M.N. Hall, "TOR complex 2: a signaling pathway of its own", Trends in Biochemical Sciences, vol. 34, pp. 620-627, 2009. http://dx.doi.org/10.1016/j.tibs.2009.09.004
- S. Aronova, K. Wedaman, P.A. Aronov, K. Fontes, K. Ramos, B.D. Hammock, and T. Powers, "Regulation of Ceramide Biosynthesis by TOR Complex 2", Cell Metabolism, vol. 7, pp. 148-158, 2008. http://dx.doi.org/10.1016/j.cmet.2007.11.015
- M. Tabuchi, A. Audhya, A.B. Parsons, C. Boone, and S.D. Emr, "The Phosphatidylinositol 4,5-Biphosphate and TORC2 Binding Proteins Slm1 and Slm2 Function in Sphingolipid Regulation", Molecular and Cellular Biology, vol. 26, pp. 5861-5875, 2006. http://dx.doi.org/10.1128/MCB.02403-05
- N.D. Bonawitz, M. Chatenay-Lapointe, Y. Pan, and G.S. Shadel, "Reduced TOR Signaling Extends Chronological Life Span via Increased Respiration and Upregulation of Mitochondrial Gene Expression", Cell Metabolism, vol. 5, pp. 265-277, 2007. http://dx.doi.org/10.1016/j.cmet.2007.02.009
- Y. Pan, E. Schroeder, A. Ocampo, A. Barrientos, and G. Shadel, "Regulation of Yeast Chronological Life Span by TORC1 via Adaptive Mitochondrial ROS Signaling", Cell Metabolism, vol. 13, pp. 668-678, 2011. http://dx.doi.org/10.1016/j.cmet.2011.03.018
- R.W. Powers, M. Kaeberlein, S.D. Caldwell, B.K. Kennedy, and S. Fields, "Extension of chronological life span in yeast by decreased TOR pathway signaling", Genes & Development, vol. 20, pp. 174-184, 2006. http://dx.doi.org/10.1101/gad.1381406
- M. Kaeberlein, and B.K. Kennedy, "Hot topics in aging research: protein translation and TOR signaling, 2010", Aging Cell, vol. 10, pp. 185-190, 2011. http://dx.doi.org/10.1111/j.1474-9726.2010.00665.x
- V. Wanke, I. Pedruzzi, E. Cameroni, F. Dubouloz, and C. De Virgilio, "Regulation of G0 entry by the Pho80–Pho85 cyclin–CDK complex", The EMBO Journal, vol. 24, pp. 4271-4278, 2005. http://dx.doi.org/10.1038/sj.emboj.7600889
- M. Wei, P. Fabrizio, J. Hu, H. Ge, C. Cheng, L. Li, and V.D. Longo, "Life Span Extension by Calorie Restriction Depends on Rim15 and Transcription Factors Downstream of Ras/PKA, Tor, and Sch9", PLoS Genetics, vol. 4, pp. e13, 2008. http://dx.doi.org/10.1371/journal.pgen.0040013
- A. Komeili, K.P. Wedaman, E.K. O'Shea, and T. Powers, "Mechanism of Metabolic Control", The Journal of Cell Biology, vol. 151, pp. 863-878, 2000. http://dx.doi.org/10.1083/jcb.151.4.863
- I. Dilova, S. Aronova, J.C. Chen, and T. Powers, "Tor Signaling and Nutrient-based Signals Converge on Mks1p Phosphorylation to Regulate Expression of Rtg1p·Rtg3p-dependent Target Genes", Journal of Biological Chemistry, vol. 279, pp. 46527-46535, 2004. http://dx.doi.org/10.1074/jbc.M409012200
- Z. Liu, and R.A. Butow, "Mitochondrial Retrograde Signaling", Annual Review of Genetics, vol. 40, pp. 159-185, 2006. http://dx.doi.org/10.1146/annurev.genet.40.110405.090613
- T. Beck, and M.N. Hall, "The TOR signalling pathway controls nuclear localization of nutrient-regulated transcription factors", Nature, vol. 402, pp. 689-692, 1999. http://dx.doi.org/10.1038/45287
- K. Voordeckers, M. Kimpe, S. Haesendonckx, W. Louwet, M. Versele, and J.M. Thevelein, "Yeast 3-Phosphoinositide-dependent Protein Kinase-1 (PDK1) Orthologs Pkh1–3 Differentially Regulate Phosphorylation of Protein Kinase A (PKA) and the Protein Kinase B (PKB)/S6K Ortholog Sch9", Journal of Biological Chemistry, vol. 286, pp. 22017-22027, 2011. http://dx.doi.org/10.1074/jbc.M110.200071
- C.J. Di Como, and K.T. Arndt, "Nutrients, via the Tor proteins, stimulate the association of Tap42 with type 2A phosphatases.", Genes & Development, vol. 10, pp. 1904-1916, 1996. http://dx.doi.org/10.1101/gad.10.15.1904
- E. Jacinto, B. Guo, K.T. Arndt, T. Schmelzle, and M.N. Hall, "TIP41 Interacts with TAP42 and Negatively Regulates the TOR Signaling Pathway", Molecular Cell, vol. 8, pp. 1017-1026, 2001. http://dx.doi.org/10.1016/S1097-2765(01)00386-0
- J. Urban, A. Soulard, A. Huber, S. Lippman, D. Mukhopadhyay, O. Deloche, V. Wanke, D. Anrather, G. Ammerer, H. Riezman, J.R. Broach, C. De Virgilio, M.N. Hall, and R. Loewith, "Sch9 Is a Major Target of TORC1 in Saccharomyces cerevisiae", Molecular Cell, vol. 26, pp. 663-674, 2007. http://dx.doi.org/10.1016/j.molcel.2007.04.020
- C.R. Burtner, C.J. Murakami, B.K. Kennedy, and M. Kaeberlein, "A molecular mechanism of chronological aging in yeast", Cell Cycle, vol. 8, pp. 1256-1270, 2009. http://dx.doi.org/10.4161/cc.8.8.8287
- A. Rego, M. Costa, S.R. Chaves, N. Matmati, H. Pereira, M.J. Sousa, P. Moradas-Ferreira, Y.A. Hannun, V. Costa, and M. Côrte-Real, "Modulation of Mitochondrial Outer Membrane Permeabilization and Apoptosis by Ceramide Metabolism", PLoS ONE, vol. 7, pp. e48571, 2012. http://dx.doi.org/10.1371/journal.pone.0048571
- P. Fabrizio, F. Pozza, S.D. Pletcher, C.M. Gendron, and V.D. Longo, "Regulation of Longevity and Stress Resistance by Sch9 in Yeast", Science, vol. 292, pp. 288-290, 2001. http://dx.doi.org/10.1126/science.1059497
- Z. Wu, S.Q. Liu, and D. Huang, "Dietary Restriction Depends on Nutrient Composition to Extend Chronological Lifespan in Budding Yeast Saccharomyces cerevisiae", PLoS ONE, vol. 8, pp. e64448, 2013. http://dx.doi.org/10.1371/journal.pone.0064448
- M. Wei, P. Fabrizio, F. Madia, J. Hu, H. Ge, L.M. Li, and V.D. Longo, "Tor1/Sch9-Regulated Carbon Source Substitution Is as Effective as Calorie Restriction in Life Span Extension", PLoS Genetics, vol. 5, pp. e1000467, 2009. http://dx.doi.org/10.1371/journal.pgen.1000467
- D.G. Nicholls, "Mitochondrial membrane potential and aging", Aging Cell, vol. 3, pp. 35-40, 2003. http://dx.doi.org/10.1111/j.1474-9728.2003.00079.x
- G. Kroemer, L. Galluzzi, and C. Brenner, "Mitochondrial membrane permeabilization in cell death.", Physiological reviews, 2007. http://www.ncbi.nlm.nih.gov/pubmed/17237344
- F. Legros, A. Lombès, P. Frachon, and M. Rojo, "Mitochondrial Fusion in Human Cells Is Efficient, Requires the Inner Membrane Potential, and Is Mediated by Mitofusins", Molecular Biology of the Cell, vol. 13, pp. 4343-4354, 2002. http://dx.doi.org/10.1091/mbc.E02-06-0330
- S.A. Detmer, and D.C. Chan, "Functions and dysfunctions of mitochondrial dynamics", Nature Reviews Molecular Cell Biology, vol. 8, pp. 870-879, 2007. http://dx.doi.org/10.1038/nrm2275
- S.B. Berman, F.J. Pineda, and J.M. Hardwick, "Mitochondrial fission and fusion dynamics: the long and short of it", Cell Death & Differentiation, vol. 15, pp. 1147-1152, 2008. http://dx.doi.org/10.1038/cdd.2008.57
- J.M.A. Oliveira, "Mitochondrial Membrane Potential and Dynamics", Mitochondrial Dysfunction in Neurodegenerative Disorders, pp. 127-139, 2011. http://dx.doi.org/10.1007/978-0-85729-701-3_8
- S.W. Suzuki, J. Onodera, and Y. Ohsumi, "Starvation Induced Cell Death in Autophagy-Defective Yeast Mutants Is Caused by Mitochondria Dysfunction", PLoS ONE, vol. 6, pp. e17412, 2011. http://dx.doi.org/10.1371/journal.pone.0017412
- M.T. Aung-Htut, Y.T. Lam, Y. Lim, M. Rinnerthaler, C.L. Gelling, H. Yang, M. Breitenbach, and I.W. Dawes, "Maintenance of Mitochondrial Morphology by Autophagy and Its Role in High Glucose Effects on Chronological Lifespan ofSaccharomyces cerevisiae", Oxidative Medicine and Cellular Longevity, vol. 2013, pp. 1-13, 2013. http://dx.doi.org/10.1155/2013/636287
- J. Lee, S. Giordano, and J. Zhang, "Autophagy, mitochondria and oxidative stress: cross-talk and redox signalling", Biochemical Journal, vol. 441, pp. 523-540, 2011. http://dx.doi.org/10.1042/BJ20111451
- E.S. Seeley, M. Kato, N. Margolis, W. Wickner, and G. Eitzen, "Genomic Analysis of Homotypic Vacuole Fusion", Molecular Biology of the Cell, vol. 13, pp. 782-794, 2002. http://dx.doi.org/10.1091/mbc.01-10-0512
- A. Simm, and H. Brömme, "Reactive oxygen species (ROS) and aging: Do we need them — can we measure them — should we block them?", Signal Transduction, vol. 5, pp. 115-125, 2005. http://dx.doi.org/10.1002/sita.200400053
- M. Raftopoulou, "Mitochondrial wrinkles: the first signs of ageing?", Nature Cell Biology, vol. 7, pp. 853-853, 2005. http://dx.doi.org/10.1038/ncb0905-853
- K.C. Kregel, and H.J. Zhang, "An integrated view of oxidative stress in aging: basic mechanisms, functional effects, and pathological considerations", American Journal of Physiology-Regulatory, Integrative and Comparative Physiology, vol. 292, pp. R18-R36, 2007. http://dx.doi.org/10.1152/ajpregu.00327.2006
- S. Marchi, C. Giorgi, J.M. Suski, C. Agnoletto, A. Bononi, M. Bonora, E. De Marchi, S. Missiroli, S. Patergnani, F. Poletti, A. Rimessi, J. Duszynski, M.R. Wieckowski, and P. Pinton, "Mitochondria-Ros Crosstalk in the Control of Cell Death and Aging", Journal of Signal Transduction, vol. 2012, pp. 1-17, 2012. http://dx.doi.org/10.1155/2012/329635
- H. Kitagaki, L.A. Cowart, N. Matmati, D. Montefusco, J. Gandy, S.V. de Avalos, S.A. Novgorodov, J. Zheng, L.M. Obeid, and Y.A. Hannun, "ISC1-dependent Metabolic Adaptation Reveals an Indispensable Role for Mitochondria in Induction of Nuclear Genes during the Diauxic Shift in Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 284, pp. 10818-10830, 2009. http://dx.doi.org/10.1074/jbc.M805029200
- A. Pascual-Ahuir, and M. Proft, "The Sch9 kinase is a chromatin-associated transcriptional activator of osmostress-responsive genes", The EMBO Journal, vol. 26, pp. 3098-3108, 2007. http://dx.doi.org/10.1038/sj.emboj.7601756
- J.B. Millar, V. Buck, and M.G. Wilkinson, "Pyp1 and Pyp2 PTPases dephosphorylate an osmosensing MAP kinase controlling cell size at division in fission yeast.", Genes & Development, vol. 9, pp. 2117-2130, 1995. http://dx.doi.org/10.1101/gad.9.17.2117
- D.A. Smith, S. Nicholls, B.A. Morgan, A.J. Brown, and J. Quinn, "A Conserved Stress-activated Protein Kinase Regulates a Core Stress Response in the Human PathogenCandida albicans", Molecular Biology of the Cell, vol. 15, pp. 4179-4190, 2004. http://dx.doi.org/10.1091/mbc.E04-03-0181
- J.A. Marquez, "The Ssn6-Tup1 repressor complex of Saccharomyces cerevisiae is involved in the osmotic induction of HOG-dependent and -independent genes", The EMBO Journal, vol. 17, pp. 2543-2553, 1998. http://dx.doi.org/10.1093/emboj/17.9.2543
- V. Reiser, H. Ruis, and G. Ammerer, "Kinase Activity-dependent Nuclear Export Opposes Stress-induced Nuclear Accumulation and Retention of Hog1 Mitogen-activated Protein Kinase in the Budding YeastSaccharomyces cerevisiae", Molecular Biology of the Cell, vol. 10, pp. 1147-1161, 1999. http://dx.doi.org/10.1091/mbc.10.4.1147
- M. Rep, V. Reiser, U. Gartner, J.M. Thevelein, S. Hohmann, G. Ammerer, and H. Ruis, "Osmotic stress-induced gene expression in Saccharomyces cerevisiae requires Msn1p and the novel nuclear factor Hot1p.", Molecular and cellular biology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10409737
- M. Rep, M. Krantz, J.M. Thevelein, and S. Hohmann, "The Transcriptional Response of Saccharomyces cerevisiae to Osmotic Shock", Journal of Biological Chemistry, vol. 275, pp. 8290-8300, 2000. http://dx.doi.org/10.1074/jbc.275.12.8290
- P.M. Alepuz, A. Jovanovic, V. Reiser, and G. Ammerer, "Stress-Induced MAP Kinase Hog1 Is Part of Transcription Activation Complexes", Molecular Cell, vol. 7, pp. 767-777, 2001. http://dx.doi.org/10.1016/S1097-2765(01)00221-0
- M. Proft, "Regulation of the Sko1 transcriptional repressor by the Hog1 MAP kinase in response to osmotic stress", The EMBO Journal, vol. 20, pp. 1123-1133, 2001. http://dx.doi.org/10.1093/emboj/20.5.1123
- E.D. Nadal, L. Casadomé, and F. Posas, "Targeting the MEF2-Like Transcription Factor Smp1 by the Stress-Activated Hog1 Mitogen-Activated Protein Kinase", Molecular and Cellular Biology, vol. 23, pp. 229-237, 2003. http://dx.doi.org/10.1128/MCB.23.1.229-237.2003
- E. de Nadal, M. Zapater, P.M. Alepuz, L. Sumoy, G. Mas, and F. Posas, "The MAPK Hog1 recruits Rpd3 histone deacetylase to activate osmoresponsive genes", Nature, vol. 427, pp. 370-374, 2004. http://dx.doi.org/10.1038/nature02258
- J.T. Nickels, and J.R. Broach, "A ceramide-activated protein phosphatase mediates ceramide-induced G1 arrest of Saccharomyces cerevisiae.", Genes & Development, vol. 10, pp. 382-394, 1996. http://dx.doi.org/10.1101/gad.10.4.382
- M. Liu, C. Huang, S.R. Polu, R. Schneiter, and A. Chang, "Regulation of sphingolipid synthesis via Orm1 and Orm2 in yeast", Journal of Cell Science, 2012. http://dx.doi.org/10.1242/jcs.100578
- D.K. Breslow, "Sphingolipid Homeostasis in the Endoplasmic Reticulum and Beyond", Cold Spring Harbor Perspectives in Biology, vol. 5, pp. a013326-a013326, 2013. http://dx.doi.org/10.1101/cshperspect.a013326
- M. Shimobayashi, W. Oppliger, S. Moes, P. Jenö, and M.N. Hall, "TORC1-regulated protein kinase Npr1 phosphorylates Orm to stimulate complex sphingolipid synthesis", Molecular Biology of the Cell, vol. 24, pp. 870-881, 2013. http://dx.doi.org/10.1091/mbc.E12-10-0753
- F.M. Roelants, D.K. Breslow, A. Muir, J.S. Weissman, and J. Thorner, "Protein kinase Ypk1 phosphorylates regulatory proteins Orm1 and Orm2 to control sphingolipid homeostasis in Saccharomyces cerevisiae", Proceedings of the National Academy of Sciences, vol. 108, pp. 19222-19227, 2011. http://dx.doi.org/10.1073/pnas.1116948108
- D.K. Breslow, S.R. Collins, B. Bodenmiller, R. Aebersold, K. Simons, A. Shevchenko, C.S. Ejsing, and J.S. Weissman, "Orm family proteins mediate sphingolipid homeostasis", Nature, vol. 463, pp. 1048-1053, 2010. http://dx.doi.org/10.1038/nature08787
- B.A. Yard, L.G. Carter, K.A. Johnson, I.M. Overton, M. Dorward, H. Liu, S.A. McMahon, M. Oke, D. Puech, G.J. Barton, J.H. Naismith, and D.J. Campopiano, "The Structure of Serine Palmitoyltransferase; Gateway to Sphingolipid Biosynthesis", Journal of Molecular Biology, vol. 370, pp. 870-886, 2007. http://dx.doi.org/10.1016/j.jmb.2007.04.086
- B.J. Niles, and T. Powers, "Plasma membrane proteins Slm1 and Slm2 mediate activation of the AGC kinase Ypk1 by TORC2 and sphingolipids in S. cerevisiae", Cell Cycle, vol. 11, pp. 3745-3749, 2012. http://dx.doi.org/10.4161/cc.21752
- M. Taniguchi, K. Kitatani, T. Kondo, M. Hashimoto-Nishimura, S. Asano, A. Hayashi, S. Mitsutake, Y. Igarashi, H. Umehara, H. Takeya, J. Kigawa, and T. Okazaki, "Regulation of Autophagy and Its Associated Cell Death by “Sphingolipid Rheostat”", Journal of Biological Chemistry, vol. 287, pp. 39898-39910, 2012. http://dx.doi.org/10.1074/jbc.M112.416552
- H. Sawai, Y. Okamoto, C. Luberto, C. Mao, A. Bielawska, N. Domae, and Y.A. Hannun, "Identification of ISC1 (YER019w) as Inositol Phosphosphingolipid Phospholipase C inSaccharomyces cerevisiae", Journal of Biological Chemistry, vol. 275, pp. 39793-39798, 2000. http://dx.doi.org/10.1074/jbc.M007721200
- M. Tanigawa, A. Kihara, M. Terashima, T. Takahara, and T. Maeda, "Sphingolipids Regulate the Yeast High-Osmolarity Glycerol Response Pathway", Molecular and Cellular Biology, vol. 32, pp. 2861-2870, 2012. http://dx.doi.org/10.1128/MCB.06111-11
- C. Su, Y. Lu, and H. Liu, "Reduced TOR signaling sustains hyphal development inCandida albicansby lowering Hog1 basal activity", Molecular Biology of the Cell, vol. 24, pp. 385-397, 2013. http://dx.doi.org/10.1091/mbc.E12-06-0477
- M. Kaeberlein, A.A. Andalis, G.R. Fink, and L. Guarente, "High Osmolarity Extends Life Span in Saccharomyces cerevisiae by a Mechanism Related to Calorie Restriction", Molecular and Cellular Biology, vol. 22, pp. 8056-8066, 2002. http://dx.doi.org/10.1128/MCB.22.22.8056-8066.2002
- E. Bilsland, C. Molin, S. Swaminathan, A. Ramne, and P. Sunnerhagen, "Rck1 and Rck2 MAPKAP kinases and the HOG pathway are required for oxidative stress resistance", Molecular Microbiology, vol. 53, pp. 1743-1756, 2004. http://dx.doi.org/10.1111/j.1365-2958.2004.04238.x
- T. Maeda, A.Y. Tsai, and H. Saito, "Mutations in a protein tyrosine phosphatase gene (PTP2) and a protein serine/threonine phosphatase gene (PTC1) cause a synthetic growth defect in Saccharomyces cerevisiae.", Molecular and cellular biology, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8395005
- T. Maeda, S.M. Wurgler-Murphy, and H. Saito, "A two-component system that regulates an osmosensing MAP kinase cascade in yeast", Nature, vol. 369, pp. 242-245, 1994. http://dx.doi.org/10.1038/369242a0
- S.M. Wurgler-Murphy, T. Maeda, E.A. Witten, and H. Saito, "Regulation of the Saccharomyces cerevisiae HOG1 mitogen-activated protein kinase by the PTP2 and PTP3 protein tyrosine phosphatases.", Molecular and cellular biology, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9032256
- S. Lee, S. Park, J. Na, and Y. Kim, "Osmolarity hypersensitivity ofhog1deleted mutants is suppressed by mutation inKSS1in budding yeastSaccharomyces cerevisiae", FEMS Microbiology Letters, vol. 209, pp. 9-14, 2002. http://dx.doi.org/10.1111/j.1574-6968.2002.tb11102.x
- W. Wang, J.X. Chen, R. Liao, Q. Deng, J.J. Zhou, S. Huang, and P. Sun, "Sequential Activation of the MEK-Extracellular Signal-Regulated Kinase and MKK3/6-p38 Mitogen-Activated Protein Kinase Pathways Mediates Oncogenic ras-Induced Premature Senescence", Molecular and Cellular Biology, vol. 22, pp. 3389-3403, 2002. http://dx.doi.org/10.1128/MCB.22.10.3389-3403.2002
- Q. Deng, R. Liao, B. Wu, and P. Sun, "High Intensity ras Signaling Induces Premature Senescence by Activating p38 Pathway in Primary Human Fibroblasts", Journal of Biological Chemistry, vol. 279, pp. 1050-1059, 2004. http://dx.doi.org/10.1074/jbc.M308644200
- H. Iwasa, J. Han, and F. Ishikawa, "Mitogen‐activated protein kinase p38 defines the common senescence‐signalling pathway", Genes to Cells, vol. 8, pp. 131-144, 2003. http://dx.doi.org/10.1046/j.1365-2443.2003.00620.x
- Z. Xia, M. Dickens, J. Raingeaud, R.J. Davis, and M.E. Greenberg, "Opposing Effects of ERK and JNK-p38 MAP Kinases on Apoptosis", Science, vol. 270, pp. 1326-1331, 1995. http://dx.doi.org/10.1126/science.270.5240.1326
- S. Grethe, M.P. Ares, T. Andersson, and M. Pörn-Ares, "p38 MAPK mediates TNF-induced apoptosis in endothelial cells via phosphorylation and downregulation of Bcl-xL", Experimental Cell Research, vol. 298, pp. 632-642, 2004. http://dx.doi.org/10.1016/j.yexcr.2004.05.007
- E.A. Nalefski, and J.J. Falke, "The C2 domain calcium‐binding motif: Structural and functional diversity", Protein Science, vol. 5, pp. 2375-2390, 1996. http://dx.doi.org/10.1002/pro.5560051201
- T. Yorimitsu, S. Zaman, J.R. Broach, and D.J. Klionsky, "Protein Kinase A and Sch9 Cooperatively Regulate Induction of Autophagy inSaccharomyces cerevisiae", Molecular Biology of the Cell, vol. 18, pp. 4180-4189, 2007. http://dx.doi.org/10.1091/mbc.E07-05-0485
- P. Ferrigno, "Regulated nucleo/cytoplasmic exchange of HOG1 MAPK requires the importin beta homologs NMD5 and XPO1", The EMBO Journal, vol. 17, pp. 5606-5614, 1998. http://dx.doi.org/10.1093/emboj/17.19.5606
- U.S. Jung, A.K. Sobering, M.J. Romeo, and D.E. Levin, "Regulation of the yeast Rlm1 transcription factor by the Mpk1 cell wall integrity MAP kinase", Molecular Microbiology, vol. 46, pp. 781-789, 2002. http://dx.doi.org/10.1046/j.1365-2958.2002.03198.x
- P. Fabrizio, and V.D. Longo, "The chronological life span of Saccharomyces cerevisiae", Aging Cell, vol. 2, pp. 73-81, 2003. http://dx.doi.org/10.1046/j.1474-9728.2003.00033.x
- S.M. Conyers, and D.A. Kidwell, "Chromogenic substrates for horseradish peroxidase", Analytical Biochemistry, vol. 192, pp. 207-211, 1991. http://dx.doi.org/10.1016/0003-2697(91)90208-B
- R.O. Poyton, B. Goehring, M. Droste, K.A. Sevarino, L.A. Allen, and X. Zhao, "[8] Cytochrome-c oxidase from Saccharomyces cerevisiae", Methods in Enzymology, pp. 97-116, 1995. http://dx.doi.org/10.1016/0076-6879(95)60133-3
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
We are grateful to Dr. David E. Levin (Goldman School of Dental Medicine, Boston University, Boston, USA), Dr. Robbie Loewith (University of Geneva, Switzerland), Dr. Francesc Posas (Universitat Pompeu Fabra, Barcelona, Spain), Dr. Yusuf Hannnun (Stony Brook University, Health Science Center, Stony Brook, New York, USA) and Dr. Paula Ludovico (ICVS, Universidade do Minho, Portugal) for generously providing plasmids and other reagents used in this study and fruitful discussion. We would like to thank Catarina Leitão (AFCU, IBMC) for technical support on flow cytometry and Paula Sampaio (ALM, IBMC) for technical assistance and data treatment on fluorescence microscopy. Funding. This work was supported by FEDER (Fundo Europeu de Desenvolvimento Regional) through the program “Programa Operacional Fatores de Competitividade-COMPETE”, by FCT (Fundação para a Ciência e Tecnologia) and by "Programa Operacional Regional do Norte (ON.2 – O Novo Norte)", through the projects PEST-C/SAU/LA0002/2013-FCOMP-01-0124-FEDER-037277 and NORTE-07-0124-FEDER-000001. V.H.F.T. (SFRH/BD/72134/2010) and R.V. (SFRH/BD/48125/2008) were supported by FCT fellowships.
COPYRIGHT
© 2013

Reduced TORC1 signaling abolishes mitochondrial dysfunctions and shortened chronological lifespan of Isc1p-deficient cells by Vitor Teixeira et al. is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.








