
Reviews:
Microbial Cell, Vol. 2, No. 4, pp. 105 - 125; doi: 10.15698/mic2015.04.197
Understanding structure, function, and mutations in the mitochondrial ATP synthase

1 Department of Biochemistry and Molecular Biology, The Chicago Medical School, Rosalind Franklin University of Medicine and Science, 3333 Green Bay Road, North Chicago, IL 60064.
2 Department of Chemical Biology and Medicinal Chemistry, Eshelman School of Pharmacy, University of North Carolina, Chapel Hill, NC.
Keywords: ATP synthase, F1 ATPase, F1Fo ATP synthase, mitochondrial diseases, petite mutations, uncoupling.
Abbreviations:
mgi - mito-chondrial genome integrity,
SNPs - single nucleotide polymorphisms.
Received originally: 16/12/2014 Received in revised form: 04/03/2015
Accepted: 07/03/2015
Published: 24/03/2015
Correspondence:
David M. Mueller, Department of Biochemistry and Molecular Biology, The Chicago Medical School, Rosalind Franklin University of Medicine and Sci-ence, 3333 Green Bay Road, North Chicago, IL 60064 david.mueller@rosalindfranklin.edu
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Ting Xu, Vijayakanth Pagadala, David M. Mueller (2015). Understanding structure, function, and mutations in the mitochondrial ATP synthase. Microbial Cell 2(4): 105-125.
Abstract
The mitochondrial ATP synthase is a multimeric enzyme complex with an overall molecular weight of about 600,000 Da. The ATP synthase is a molecular motor composed of two separable parts: F1 and Fo. The F1 portion contains the catalytic sites for ATP synthesis and protrudes into the mitochondrial matrix. Fo forms a proton turbine that is embedded in the inner membrane and connected to the rotor of F1. The flux of protons flowing down a potential gradient powers the rotation of the rotor driving the synthesis of ATP. Thus, the flow of protons though Fo is coupled to the synthesis of ATP. This review will discuss the structure/function relationship in the ATP synthase as determined by biochemical, crystallographic, and genetic studies. An emphasis will be placed on linking the structure/function relationship with understanding how disease causing mutations or putative single nucleotide polymorphisms (SNPs) in genes encoding the subunits of the ATP synthase, will affect the function of the enzyme and the health of the individual. The review will start by summarizing the current understanding of the subunit composition of the enzyme and the role of the subunits followed by a discussion on known mutations and their effect on the activity of the ATP synthase. The review will conclude with a summary of mutations in genes encoding subunits of the ATP synthase that are known to be responsible for human disease, and a brief discussion on SNPs.
INTRODUCTION
Peter Mitchell first proposed that the proton potential was used to provide the energy needed for the synthesis of ATP. While the chemiosmotic hypothesis [1] was proposed in 1961, because of the complexities of the enzyme, we are just now understanding the molecular details of ATP synthesis. The ATP synthase is a large multi-subunit enzyme complex composed of up to 20 different subunits or polypeptides that are in some cases present in multiple copies. The ATP synthase is bound to the inner membrane of the mitochondrion with a number of subunits being water-soluble while others are integral membrane proteins.
–
The mitochondrial ATP synthase is composed of two separable components: F1 (factor 1) and Fo (factor that confers sensitivity to oligomycin). The ATP synthase is a reversible molecular motor comprised of two parts: a proton turbine (within Fo) and a molecular machine (F1) that uses rotational energy to form ATP from ADP and phosphate. The proton turbine is powered by the flow of protons down a potential gradient across the mitochondrial membrane created by the electron transport chain during respiration. The rotor of the turbine is within the F1 and when it rotates, drives the synthesis of ATP in the general mechanism described by the binding change hypothesis of Paul Boyer [2].
–
The ATP synthase acts to convert the energy of oxidation-reduction reactions of the electron transport chain (respiration) to the phosphorylation of ADP. The synthesis of ATP is “coupled” to the respiratory chain via the proton potential. The number of ATP molecules made, per number of atoms of oxygen reduced in the electron transport chain, (P/O ratio) is a measure of the coupling of ATP synthase with the electron transport chain [3]. The P/O ratio is dependent upon a number variables, including the membrane integrity, site of the electron transport chain where the electrons are accepted, and the integrity of the ATP synthase. Because of these variables and basic experimental variations, a range of P/O have been reported, but the consensus values for mammalian mitochondria, are 2.5 and 1.5, for electrons entering at NADH dehydrogenase and succinate dehydrogenase, respectively [4][5]. The recent understandings of the structure/function relationship provide insight into the coupling capacity of the ATP synthase partly based upon the theoretical number of ATP molecules made per flux of protons. Using the c-ring stoichiometry of 8 (see discussion on Fo below) for the bovine enzyme, the maximal theoretical P/O ratio for mammalian mitochondria is 2.7 for NADH and 1.6 for succinate [3]. Yeast S. cerevisiae lacks the first coupling site [6] and considering the yeast c-ring stoichiometry of 10, the P/O ratio for NADH and succinate is about 1.3. However, for analysis of the ATP synthase function, given that all variables outside the ATP synthase are equal, a higher P/O ratio corresponds to a higher efficiency of the enzyme. Classical chemical uncouplers, such as 2,4 dinitrophenol, allow respiration without ATP synthesis thus reducing the P/O ratio to zero, by dissipating the proton potential across the mitochondrial membrane. However, the flow of protons through Fo into the matrix is also coupled – coupled to the phosphorylation of ADP, and mutations can alter the coupling which reduces the P/O ratio.
STRUCTURE AND ORGANIZATION OF THE F1Fo ATP SYNTHASE
A composite model of the mitochondrial ATP synthase is shown in Figure 1. This composite model was derived from the crystal structure of the yeast F1 ATPase (pdb: 2HLD, the yeast c10-ring, 3U2F, and the bovine stator derived from two structures, 2WSS and 2CLY [7][8][9][10][11]). Table 1 lists the subunit composition of the bovine, yeast and E. coli enzymes along with the genes and relevant information on the structure/function relationship. The composite model is missing components of Fo, which provide access for protons from the matrix and intermembrane space to the proton acceptor/donor site of the c-ring, which is positioned in the center of the lipid bilayer of the inner mitochondrial membrane. Based on subunit composition, the bacterial enzyme is much less complex than the mitochondrial enzyme, which may be due to the greater need for regulation in the eukaryotic cell. However, while the details differ, the general mechanism of ATP synthesis is conserved from the bacterial to the mammalian enzyme. The structure/function relationship of the subunits will be discussed in three groups: F1, Fo, and stator.
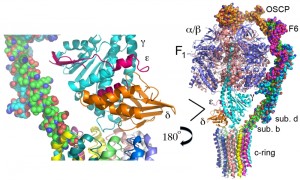 | FIGURE 1: Structural representation of the ATP synthase. A composite representation of the ATP synthase is shown on the right with the F1, subunits of the stator, and the c10-ring shown. The structure of the F1 and c10-ring were derived from the x-ray crystal structures (2HDL and 3U2F) while the structures of the stator (peripheral stalk) were derived from the x-ray crystal structures of the bovine components (2WSS and 2CLY). Note, that a number of subunits have not been represented in the structure, most notably, subunit-a, which forms the proton half-channels. |
The F1 ATPase
The mitochondrial F1 ATPase has the subunit composition α3β3γδε. The molecular weight of the subunits are 55 kDa, 51 kDa, 30 kDa, 15 kDa, and 5.7 kDa for the mature bovine α, β, γ, δ, and ε subunits, respectively. The X-ray crystal structure of the bovine F1-ATPase provided the first look at the structure/function relationship of the subunits in the F1 ATPase [12] and subsequent structures, some with bound inhibitors, have given greater understanding into the molecular mechanism of ATP synthesis [13][14][15][16][17][18][19][20][21][22][23]. The α- and β-subunits share only about 20% identity but they form nearly identical folds and the α-carbon atom positions differ only by about 2.8 Å rmsd (root mean square deviation) with the greatest spatial divergence in the last 40 residues. The nucleotide binding sites are at the interfaces between the α- and β-subunits with the catalytic site formed primarily by the β-subunit and the non-catalytic site formed primarily by the α-subunit. In accordance, there are 6 nucleotide-binding sites in the α3β3γ assembly with 3 being catalytic binding sites and 3 being non-catalytic binding sites. Support for the residues involved in the binding and catalysis deduced from the crystal structure was obtained by extensive mutagenesis of primarily the E. coli enzyme, prior to and after the structure determination (for review, see [24]). The P-loop (residues 156-163) is integral to the catalytic site with β-subunit residues Gly161/Lys162/Thr163 being nearly invariable with only Ser163 being a functional variant [25]. For ATP hydrolysis, β-Glu188 has been concluded to act as a catalytic base that activates a water molecule in a reaction mechanism that has been described as SN2 [26] where in the transition state, the γ-phosphate assumes a trigonal-bipyramidal arrangement of the oxygen atoms. ATP hydrolysis is the reverse of ATP synthesis and microscopic reversibility is widely accepted for this reaction. β-Asp256 and the β- and γ-phosphates for ATP form the Mg2+ binding site. Arg373 of the α-subunit is essential for catalysis and the crystal structure suggests that it is involved in stabilizing the penta-coordinate transition state. This role was more clearly demonstrated in the structure of the bovine F1 ATPase with the transition state analogue, ADP:AlF4, which showed the guanidinium group of α-Arg373 in a position to stabilize the transition state [18][27]. In the structure of the yeast F1 ATPase, α-Arg373 is also shown to participate in the binding of inorganic phosphate, one of the substrates for the synthesis of ATP [7].
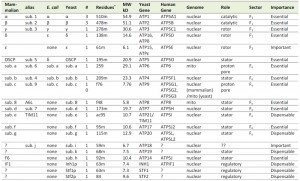 | TABLE 1. f: N-formyl Met, ac: putative acylation, m: mature; +The N-terminus of the mature subunit was derived by many investigators, but a more comprehensive list by Arnold et al., [28]. |
The crystal structure of the F1 gives clues as to why the non-catalytic sites are catalytically inactive. The relative position of the nucleotide bound in the catalytic and non-catalytic sites are nearly identical suggesting that the nucleotide-binding site is conserved. In the non-catalytic site, β-Arg356 is replaced by α-Arg373, α-Asp269 replaces β-Asp256, and α-Gly174/α-Lys175/α-Thr176 are replaced by β-Gly161/β-Lys162/β-Thr163. However, β-Glu188 is replaced by α-Gln208 thereby removing the carboxylate that would otherwise act as a catalytic base. In addition, as will be discussed below, the structure of the catalytic sites alternate between a closed and open conformation during the catalytic cycle but all three of the non-catalytic sites are in the closed conformation. The absence of a catalytic base and the inability to transition between different catalytic conformations, as evidenced by the absence of open conformation, are major determinants in the catalytic incapacity of the non-catalytic sites.
–
The seminal x-ray structure of the bovine F1 ATPase supported a rotational mechanism for ATP synthesis [12]. The three catalytic sites were different in both structure and occupancy; sites were occupied with AMP-PNP, ADP, and the third catalytic site had no bound nucleotide (empty). As such, the active sites were named, TP, DP, and E and the corresponding β-subunits were named, βTP, βDP and βE. The structure of the yeast F1 ATPase showed very similar results but the DP site was occupied with AMP-PNP [7]. The structure of the nucleotide-binding site in E was open and largely different from either DP or TP and this explained why no nucleotide was bound to βE. For instance, α-Arg373 is displaced 7.2 Å in the E site as compared to the TP or DP site and is not in position to interact with bound nucleotide. In the structure of the yeast F1 ATPase, inorganic phosphate (or possibly sulfate) was bound in βE supporting the conclusion that βE is a catalytically important state [7]. The asymmetry of the catalytic sites is determined by the position of the γ-subunit, which is located in the center of the α3β3 core. The structure suggested that the conformation of the catalytic site is determined by the position of the γ-subunit, which rotates relative to the α3β3 core in 120o increments during the catalytic cycle. Indeed, this was later proven by direct visualization of the movement of actin filaments fixed to the γ-subunit of bacterial F1 ATPase due to hydrolysis and subsequently by demonstration of ATP synthesis driven by physical rotation of the rotor [29][30][31]. This mechanism was also consistent with the binding change site hypothesis for ATP synthesis proposed by Boyer [2]. Thus, the γ-subunit is a rotor that rotates within the α3β3 catalytic core effecting conformational changes in the nucleotide binding sites thereby effecting ATP synthesis.
–
The molecular details of the motor movement of the synthetic pathway for ATP synthesis have been largely studied by determining the hydrolytic pathway with the assumption of microscopic reversibility. Single molecule experiments have mostly been done using the F1 from Bacillus PS3 ([29][32][33][34][35][36][37][38][39][40][41]) however, experiments using enzyme from E. coli, yeast, and human have also been reported [36][38][39][40][41][42]. The results using F1 from Bacillus, E. coli, and yeast generally agree, but the details of the mechanism differ slightly from that of the human enzyme. Figure 2 shows a molecular mechanism for the synthesis of ATP based on the studies for the hydrolysis of ATP by the bacterial and human enzymes [42] and also based on the structure of the bovine enzyme [12]. For the bacterial and yeast enzymes, there are 2 dwell positions during ATP hydrolysis, which can vary slightly: one at 0o, which corresponds to ATP binding and one at 80-90o during the catalytic reaction. For the Bacillus PS3 enzyme, Pi release also occurs at 80o. For the human enzyme, there are 3 dwell positions during ATP hydrolysis: at 0o due to ATP binding, at 65o due to Pi release, and at 90o due to ATP hydrolysis. The steps occur 3 times for a full rotation and thus each step is repeated in 120o increments.
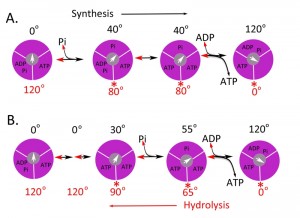 | FIGURE 2: Possible scheme for site occupancy during ATP synthesis. Shown is the one possible mechanism for ATP synthesis based on the data for the bacterial [35] and yeast [36] enzyme (A) and the human (B) F1 ATPase [42]. The direction from left to right is for ATP synthesis and right to left for ATP hydrolysis. The three binding sites are shown in magenta and the γ-subunit in grey with the arrow indicating the relative position. The asterisks (*) indicate the dwell angle observed during ATP hydrolysis. |
In terms of ATP synthesis for the human enzyme, at 0o, F1 is proposed to contain ADP and Pi in one site and ATP in a second site, while the third site is empty. The γ-subunit rotates by 30o, which is followed by the phosphorylation of ADP to ATP. Pi binds to the empty subunit followed by the rotation of the γ-subunit by 25o. The final step consists of the binding of ADP, the rotation the γ-subunit by 65o and the release of ATP made in a prior round. In this scheme, the newly bound ADP and Pi are not converted to ATP until the second round, and the newly formed ATP is not released until the third round. This mechanism fits nicely with the biochemical and structural properties of the enzyme. The three sites identified by crystallography of the bovine enzyme [12], TP, DP, and E sites, relate to the ground state structure likely at about 65o in this scheme. The conversion of ADP and Pi to ATP has been shown to be isoenergetic by both O18 exchange studies and hydrolysis of ATP under unisite conditions [2][43][44][45][46]. During ATP hydrolysis, there are 2 steps that provide torque: the release of Pi and ATP binding [32][35][47]. In the synthesis direction, energy would be needed for the binding of Pi and the release of ATP. In this scheme, all three sites are cooperating in the synthesis of ATP; one site is making ATP, the second site is binding ADP and Pi, and the third site is releasing ATP.
–
This mechanistic scheme predicts that the inhibition of one of the three catalytic sites will completely inhibit the enzyme. To assess this, the Bacilus PS3 enzyme was prepared, containing 1, 2 and 3 β-subunits with the catalytic base mutation, β-E190Q [48]. ATPase activity of the enzyme with just one of three β-catalytic sites mutated was completely inhibited confirming that all three catalytic sites must be functional for activity. A similar study was done using mutations (“slow”) in the β-subunit that reduced the binding constant for ATP to the catalytic site [49]. In this case, slowing the binding of ATP at one site had no effect on the rate of ATP binding at the other 2 sites. At a molecular level, the β-E190Q mutation acted in a dominant manner, while the “slow” mutations acted in a co-dominant manner. These results have implications in understanding the genetics of disease causing mutations in the α- and β-subunits of the ATP synthase.
–
The rotation of the γ-subunit within the core of the F1 ATPase causes the conversion of the conformation of the active site from E to DP to TP. There are 2 primary sites of interaction between the β-subunit, which largely forms the active site, and the γ-subunit, which forms the rotor and these regions are referred to as Catch 1 and Catch 2. Catch 1 includes residues in a loop formed by residues 312-319 in the β-subunit and residues in a helix of the γ-subunit formed by residues 252-255. Catch 2 encompasses residues within a loop from 387-398 in the β-subunit and residues 87-90 and 235-242 in the γ-subunits [12].
–
The rotor, also called the central stalk, is composed of the γ-subunit along with the δ- and ε-subunits. The structures of the δ- and ε-subunits were not entirely visible in the first structure of the bovine F1 ATPase but became clear in the structure of the bovine F1 ATPase inhibited with N,N’ dicyclohexylcarbodiimide (DCCD) [17]. The belief is that the inhibition by DCCD was incidental to the resolution of the δ- and ε-subunits and not causative. The δ- and ε-subunits are positioned at the base of the rotor and appear to sit close to the Fo portion of the ATP synthase. As is discussed below, mutagenesis experiments suggest that the γ-, δ- and ε-subunits share a common function in enzyme coupling – i.e., coupling proton translocation though Fo with rotation of the rotor.
–
Factor Fo
Factor Fo is also commonly referred to as F0 (F zero). Efraim Racker named factor Fo for the factor that conferred sensitivity of the F1 ATPase to the antibiotic, oligomycin [50][51][52]. Oligomycin is now known to bind to subunit-c of Fo and prevent proton flow. Recently, the structure of oligomycin bound to yeast subunit c has been determined by X-ray crystallography at 1.9 Å resolution providing molecular details into oligomycin binding and an understanding of how oligomycin blocks proton translocation [53].
–
Based on the structural and genetic data, it is postulated that oligomycin binds on the face of the c-ring at the region comprised of the proton half channel, formed in part by subunit-a [53]. The Fo sector is defined by the number of subunits in the biochemical preparation, all of which have the common characteristic of being membrane proteins or tightly associated with the membrane proteins. The Fo portion of the ATP synthase comprises the proton turbine as well as the base of the stator, while the rotor is composed of γδε [54]. The core of Fo is composed of an oligomer of the c-subunits, which contain an essential carboxylate from the side chain of either a glutamate or aspartate residue. The side chain carboxyl acts as the proton donor and acceptor in proton translocation pathway. Two half-channels (or pathways) are postulated to exist and are thought to be formed by subunits-a, which provide access to the essential carboxylate on subunit-c and to the aqueous phase of the matrix and intermembrane spaces of the mitochondrion [55][56][57]. Other subunits, such as A6L, may either modulate or provide structural support for the Fo portion of the enzyme. A recent model of the bovine ATP synthase using cryo-electron microscopy was able to show the arrangement of subunits a, b, c, e, f, g, and A6L, within the membrane domain [58].
–
The number of c-subunits varies between species with known stoichiometries ranging from 8-15 [3][59][60][61][62][63]. Bovine Fo is composed of a c8-ring [3] while yeast Fo is composed of a c10-ring [8][59]. The c-ring rotates within the membrane in steps determined by the number of c-subunits in the c-ring. As such, movement of 8 protons are used to rotate the bovine c-ring 360o driving the synthesis of 3 ATPs. This analysis provides insight into the thermodynamic considerations for ATP synthesis and provides theoretical values for ATP/H+ ratios and therefore, P/O ratios [3].
–
The essential glutamate is located in the center of helix 2 of the c-ring subunit, which positions it in the middle of the lipid bilayer. Thermodynamic considerations suggest that only the protonated form of glutamate is able to exist in the center of the bilayer and protonation/deprotonation reaction must occur in a hydrophilic environment created by two half-channels. The half-channels provide access to either the matrix or the intermembrane space thereby allowing proton flow between the compartments. The half-channels are thought to be comprised of subunit-a, but may also include subunit b and other components of Fo. There is only one strictly conserved residue in subunit-a, Arg210 (E. coli numbering, Arg259, human), which is believed to be a principle component in the proton translocation pathway (see [64] for review). Recent electron microscopy images of the Thermus thermophilis V-ATPase and the bovine ATP synthase suggest that the contact between Fo and the half-channel is surprisingly small [58][65]. This loose association may be essential for the high-speed rotation of the c-ring.
–
The stator
The stator is also referred to as the peripheral or extrinsic stalk. The stator functions to hold F1 fixed to allow rotation of the rotor within the core of F1. The stator provides a structural support and is not involved directly in the catalytic reaction. The stator is composed of the oligomycin sensitive conferring protein (OSCP, subunit 5), subunit b, subunit d, and F6 and the X-ray structure has been solved for the peripheral stalk of bovine enzyme [9][11]. Subunit b is anchored to the membrane while OSCP is locked to the top of F1. (The naming of subunit 5 as OSCP is unfortunate since OSCP does not participate in forming the oligomycin-binding site.) OSCP was so named because oligomycin prevents proton flow through Fo and inhibits ATP hydrolysis only if F1 is functionally attached and coupled to proton movement. Breaking the stator uncouples ATP hydrolysis from proton translocation because the F1 core can spin instead of the rotor. The primary structure of the stator proteins shows low conservation between bovine and yeast, as evidenced by yeast subunit h and bovine F6, which have just 14.5% sequence identity. However, the function is conserved because expression of a cDNA encoding bovine F6 complements the deletion mutation of the gene encoding subunit h in yeast [66].
–
Other subunits
Subunit f is a component of the bovine and yeast ATP synthase [54][67][68], however, the role is uncertain. Subunit f associates with Fo and deletion of the gene in yeast results in phenotypes that are typical for an ATP synthase that is uncoupled [68] (see below). The role of subunit f is still ambiguous as loss of either a component of the peripheral stalk or Fo give similar phenotypes. Yeast with a null mutation in the corresponding gene is unable to grow on a nonfermentable carbon source and loses mitochondrial DNA at a high rate [68]. As such, subunit f is an essential component of the mitochondrial ATP synthase.
–
Subunit i (aka subunit j) was identified as a component of the yeast ATP synthase nearly coincidently by two laboratories, which is why it has an alias [69][70]. A mammalian homologue for subunit i has not been identified though this does not exclude its presence. Subunit i easily dissociates from the ATP synthase complex, which explains why it had not been identified in prior studies. Deletion of the gene encoding subunit i gave different results in the two laboratories. In one case, deletion of the gene encoding yeast subunit i does not alter the assembly of the ATP synthase; the resulting ATP synthase is functional, but is less well coupled based on the P/O ratio and growth phenotype of the yeast [69]. However, in the second laboratory, deletion of the gene encoding subunit i resulted in complete loss of the ATP synthase with absence of the mitochondrial encoded subunits [70]. These results are not as different as they appear because, a loss of coupling of the ATP synthase will result in the loss of mitochondrial DNA [71]. As such, these results differ by a matter of degree, which can be caused by different growth conditions or genetic background of the yeast strains. While the role of subunit i is uncertain, it does appear to provide a role in the efficient coupling of the yeast ATP synthase.
–
A dimer form of the ATP synthase has been shown in yeast [28] and there is evidence of dimer forms of the ATP synthase in bovine, chloroplast, and other species of fungi [72][73]. The dimer form appears to be involved in formation of cristae of the inner mitochondrial membrane, which may be why the dimer form has not been shown for the bacterial enzyme [74][75]. Subunits e, k, and g are selectively associated but only subunits e and g are necessary for dimer formation [28][75]. Subunit k has not been identified in the mammalian ATP synthase although this does not exclude the possibility of a mammalian homolog. Deletion of the gene encoding subunits e or g has pleiotropic effects including a decrease of cytochrome oxidase activity [76][77]. While it is not uncommon for mutations in genes encoding mitochondrial proteins to exhibit pleiotropic effects, this effect was unusual in that the levels of cytochromes a + a3, c + c1, and b were unaltered suggesting that activity could be altered without altering synthesis or stability of the cytochromes. This and an unrelated study [78] suggest a link, which will be discussed later, between the ATP synthase and the respiratory chain.
–
The natural inhibitor protein, IF1, is thought to regulate the ATPase activity of the ATP synthase [79]. The inhibitor protein has a unique characteristic in that it inhibits ATP hydrolysis, but not ATP synthesis. In yeast, there are also related molecules, Stf1p and Stf2p. Stf1p appears to stabilize the interaction of IF1 with F1 or possibly is an isoform of IF1 [80]. Deletion of the gene encoding IF1 from yeast results in mitochondria that demonstrate uncontrolled hydrolysis of ATP in the presence of a chemical uncoupler, such as 2,4 dinitrophenol [81]. A number of studies have suggested that inhibitor protein has a role in preventing hydrolysis of ATP under ischemic conditions in mammalian heart [82][83][84][85][86] and other metabolic effects [87]. While this is a conceptually attractive hypothesis, the energy of the hydrolysis of ATP by the ATP synthase is not lost – it is coupled to the pumping of protons across the mitochondrial membrane creating a proton gradient. In contrast, the hydrolysis of ATP by free F1 ATPase is futile and wasteful, and it would be important for the cell to control this hydrolysis. Free F1 ATPase has been suggested to be an intermediate in the assembly pathway of the ATP synthase [88]. Thus, IF1 may serve a role in preventing the hydrolysis of ATP by free F1 that occurs during biogenesis of the ATP synthase complex. Stf2p appears to be important in yeast during times of stress or dehydration [89]. Interestingly, over-expression of Stf2p in yeast results in a reduction of reactive oxygen species (ROS) in response to stress and thus provides a possible target to reduce ROS production in humans. In this regard, IF1 has been shown to be important for the survival of HeLa cells when they are exposed to high concentrations of ROS, but IF1 is not important for cell survival under normal conditions [90].
–
The X-ray crystal structure of the bovine inhibitor protein bound to the bovine F1 ATPase has shown that IF1 is trapped between the pair of the α- and β-subunits, which form the DP site [91][92]. The mechanism of inhibition of IF1 was described in terms of the binding change hypothesis, but the explanation will be given in terms of the nomenclature used for the active sites of the ATP synthase. IF1 is postulated to initially bind to the open site (E), which closes to form the TP:IF1 site during ATP hydrolysis. A second round of ATP binding and hydrolysis converts the TP:IF1 site to the DP:IF1 site where it is locked in the final inhibited form. IF1 is thought to prevent the conversion of the DP:IF1 site to the E:IF1 site by steric interference within the interface of the α- and β-subunits. The reason as to why IF1 is unable to inhibit the reverse reaction, ATP synthesis, is still uncertain. The structural studies [91] do not support its role as a ratchet, which has been proposed for the inhibitory mechanism of the bacterial ε-subunit [93]. The trivial explanation is that the conditions, such as pH and ΔΨ, during ATP synthase differ from conditions under which ATP hydrolysis occur, and IF1 does not bind to F1 under synthesis conditions.
–
Deletion of genes encoding F1 subunits in yeast
A mutation that completely eliminates expression of a gene – a null mutation – is the simplest mutation to understand both at the level of the structure/function relationship of the protein and in relation to clinical relevance of the mutation. However, there are at least 2 instances with added clinical relevance and understanding of structure/function of the protein of the null mutation. First, haploinsufficiency is the situation where loss of one of the wild type alleles in a diploid organism causes a disease. Thus, a 50% drop in expression level is sufficient to cause disease or defective phenotype. Second is the situation where a null mutation causes a dominant negative or gain of (dysfunctional) activity. Both of these cases appear to be applicable in the yeast model system of null mutations in genes encoding subunits of the ATP synthase as well as for the yeast vacuolar ATPase [94][95].
–
All five subunits of the mitochondrial F1 ATPase are essential for the function of the ATP synthase. A priori, it would be predicted that deletion of any one of the five genes encoding the subunits of F1 would result in similar, if not identical, phenotypes. However, the situation is much more complicated than predicted [71]. The phenotypes of the yeast strain with a deletion mutation in either the gene encoding α or β, were typical and not surprising; the cells were defective in oxidative phosphorylation and did not grow on medium with a nonfermentable carbon source. Different from the phenotypes caused by loss of α or β, yeast cells with a deletion of the gene encoding γ result in very small colonies and the cells had completely lost their intact mitochondrial DNA, i.e., cytoplasmic petite (ρ–/ρ0). However, deletion of genes encoding α and γ (ΔαΔγ) in the same cell results in cells with a phenotype that is similar to that of yeast with a deletion mutation just in the gene encoding α (Δα). Similarly, cells with a deletion mutation in the gene encoding β had the same phenotype as cells with double mutation, ΔβΔγ. Deletion of the gene encoding δ from yeast resulted in a phenotype that was similar to those obtained with deletion of the gene encoding γ, but not as dramatic. Similar to cells that have a deletion in the gene encoding the γ-subunit, cells with a deletion of the gene encoding δ caused the cells to lose 100% of their intact mitochondrial DNA. Lastly, deletion of the gene encoding ε had the weakest effect on the phenotype, but 60% of the cells became cytoplasmically petite. In each case, the percentage of cells that became cytoplasmically petite returned to normal level when a null mutation in the gene encoding either α or β was added. This effect indicates that the phenotype caused by the deletion of the gene encoding either α or β, was epistatic to that caused by deletion of the gene encoding either the γ, δ or ε-subunit.
–
Mutations in genes encoding chaperones, ATP11 or ATP12, had the same epistatic effect, in respect to mutations in the genes encoding the γ-, δ-, and ε-subunits, as deletion mutations in the genes encoding α or β. Atp11p and Atp12p are needed for the assembly of the α- and β-subunits into F1 and yeast strains with mutations in either ATP11 or ATP12 have phenotypes similar to yeast strains with mutations in the genes encoding the α and β-subunits [96][97][98][99][100][101]. A reasonable explanation for the effect was that a subcomplex of the ATP synthase could assemble without one of the rotor subunits, but not without α or β, and the subcomplexes devoid of a rotor subunit were responsible for the additional phenotypes.
–
The effect by the loss of a subunit in the rotor can be deduced after considering the role of the rotor. With loss of the rotor, one would predict that the F1Fo subcomplex would allow protons to flow through Fo without Fo being coupled to the rotation of the rotor. Under this condition, the enzyme would be completely uncoupled and the mitochondria would exhibit a P/O ratio of zero. However, cells lacking γ or δ become 100% cytoplasmically petite. Since Fo subunits are encoded in the mitochondrial DNA, it is impossible to assess defects in coupling when the strain becomes entirely petite. Two different approaches helped resolve this problem. First, diploid yeast cells that were heterozygote for a mutation in the gene encoding γ or δ were impaired, but not totally defective in oxidative phosphorylation. Analysis of mitochondria isolated from these strains indicated that the deletion mutations caused an uncoupling of the ATP synthase presumably due to a leakage of protons into the mitochondria via the uncoupled ATP synthase subcomplex [102]. The second approach used a doxycycline-regulated expression system to gradually reduce the level of δ in the cell [103]. Using this system, direct evidence was obtained that the ATP synthase could assemble in the absence of δ and that this δ-less ATP synthase subcomplex was uncoupled because of a Fo mediated proton leak.
–
A central question that was left unanswered in this study was the relationship between uncoupling and loss of mitochondrial DNA, i.e. cytoplasmic petite formation. While this has not been conclusively answered, there is a reasonable explanation that is consistent with what is known about mitochondrion biogenesis [104]. The biogenesis of the mitochondrion is dependent on the import and processing of proteins that are encoded in the nucleus and made in the cytoplasm. The import of the mitochondrial precursor proteins from the cytoplasm into the mitochondrion is dependent on proton potential across the mitochondrial membrane [105][106]. (In petite strains or under anaerobic conditions, the mitochondrial membrane potential is generated by the electrogenic exchange of cytoplasmic ATP4- with matrix ADP3‑ [107][108][109].) As such, uncoupling of the mitochondrial membrane will prevent the import of newly synthesized proteins and thus inhibit mitochondrion biogenesis. While yeast is a facultative anaerobe and able to survive without oxidative phosphorylation, all eukaryotic cells require mitochondrion and thus inhibiting the biogenesis is lethal. If the ATP synthase is uncoupled, the cell can only survive if the proton leak is blocked and that is most easily achieved in yeast by eliminating the mitochondrial DNA, which encodes subunits a, c and 8 which comprise the proton pathway. Thus, there is a correlation between the degree of uncoupling of the ATP synthase and the propensity of the cells to become cytoplasmic petite.
–
The cellular phenotypes and biochemical consequences associated with deletion of genes encoding α, β, γ, δ, and ε, provided key insights into role of the δ and ε subunits in the coupling of the enzyme. The deletion mutations in the genes encoding the γ, δ, or ε subunits resulted in an enzyme that was uncoupled and as such, provides a basis for recognizing missense mutations that uncouple the ATP synthase. Even the loss of a single copy of the genes encoding the γ or δ-subunits in a diploid strain can result in a dysfunctional uncoupling activity, i.e., haploinsufficiency [102].
–
The mitochondrial ε-subunit is unique in that it is the only F1 subunit for which there is no bacterial homolog. As with deletion of the gene encoding subunit i, deletion of the yeast gene encoding ε has given different phenotypes depending on the laboratories; in one case the loss of ε inactivated the enzyme while in the other case, its loss severely compromised the enzyme based on the growth phenotype on a non-fermentable carbon source [110][111]. This discrepancy has been apparently resolved by the identification of selective mutations in the genes encoding either subunit a and c, which suppressed the growth phenotype in the yeast strain with the null mutation in the gene encoding the ε-subunit [112]. It was suggested that the strains with a null mutation in the gene encoding the ε-subunit that were able to grow on a nonfermentable carbon source, had picked up a suppressor mutation in either the gene encoding subunit a or c.
–
Missense mutations that uncouple the ATP synthase
The coupling of the ATP synthase is defined here as the coupling of ATP synthesis or hydrolysis with the movement or pumping of protons. Mutations that inhibit activity of the ATP synthase can act either by inhibiting the enzymatic reaction, as in an active site mutation, or by uncoupling proton movement with the reaction cycle. The coupling of rotation of the rotor with flow of proton and the synthesis of ATP is an intricate mechanism caused by numerous inter-subunit contacts. As such, mutations exist that alter these interactions and affect coupling. A genetic selection scheme, seemingly unrelated to the ATP synthase, selected cells with mutations in genes encoding the ATP synthase and these mutations cause an uncoupling of the ATP synthase.
–
Cytoplasmic “petite” yeast strains have either lost large parts of, or completely lost their mitochondrial DNA, rendering the yeast incompetent for synthesis of mitochondrial encoded proteins. The yeast, Kluyveromyces lactis, is a facultative anaerobe like Saccharomyces cerevisiae, but differs in that it requires mitochondrial DNA even when grown on a fermentable carbon source. Based on this phenotype, the cells are referred to as being “petite negative”. This is in contrast to S. cerevisiae, which is petite positive as deletions in the mitochondrial DNA are not lethal. Mutations in K. lactis were identified that convert the cells from petite negative to petite positive and the complementation group was named mitochondrial genome integrity (mgi) [113][114][115][116][117][118]. Surprisingly, the mgi mutations mapped in the genes encoding α-, β-, and γ-subunits of the mitochondrial ATP synthase. The yme1 mutation converts S. cerevisiae from petite positive to petite negative. A number of mutations were isolated from a yme1 S. cerevisiae strain that converted the yeast from petite negative, back to petite positive [119][120]. These mutations also mapped to the genes encoding the ATP synthase with some of them being identical to mgi mutations identified in K. lactis. In a manner analogous to K. lactis, the survival of the blood stream form of Trypanosoma brucei requires mitochondrial gene expression despite the absence of oxidative phosphorylation during this stage of the life cycle. However, a single polymorphism in the gene encoding the γ-subunit, which mapped in the same region as the mgi mutations in the gene encoding the γ-subunit, was identified that allowed for dyskinetoplasty (lack of mitochondrial DNA) in T. brucei. [121][122]. These diverse systems converging on common mutations, suggest a shared mechanism linking the ATP synthase with loss or retention of mitochondrial DNA.
–
Mutations analogous to mgi mutations were made in yeast S. cerevisiae and the biochemistry and structure of the enzyme was studied [123]. S. cerevisiae is petite positive and when mgi mutations are introduced into this yeast there is a striking increase in the percentage of petite strains during normal division. A variety of biochemical studies on the isolated mitochondria indicated that the mgi mutations caused an uncoupling of the ATP synthase presumably caused by proton leakage through Fo. Mapping the mutations on the structure of the yeast F1 ATPase indicated that the mutations clustered in two regions of the F1
–
ATPase – along the collar region, which steadies the rotation of the central stalk and in the region first identified in bovine F1 ATPase and referred to as Catch 2 region [12] in the β-subunit which interacts with the γ-subunit (Figure 3) [123]. X-ray crystallography of some of the mutant structures provided a structural basis for the mechanism of uncoupling rotation of the rotor from ATP hydrolysis or synthesis [124]. Interesting, two of the mgi mutations (α-Asn67Ile and β-Val279Phe) appear to disrupt phosphate binding to the E site which, if correct, would suggest that these mutations allow for proton driven rotation in the absence of substrate (phosphate) being bound thereby resulting in a less efficient, less coupled, enzyme.
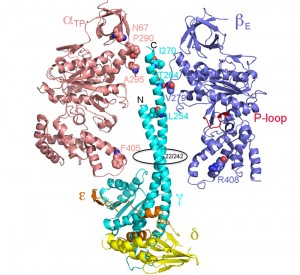 | FIGURE 3: Location of residues in the yeast ATP synthase classified as mgi. The structural representation is derived from the x-ray structure of the yeast F1 ATPase (2HDL). Shown are the αβγδε subunits, as indicated. The α3β3 core has been stripped down to a single α and β subunit to simplify the view. The mgi residues are as labeled. The N- and C- terminal ends of the γ-subunit are indicated as is the region of the orifice of the α3β3 assembly, which is located at residues 22 and 242 of the yeast γ-subunit. The nucleotide binding P-loop domain is colored red and labeled. |
While the structural and biochemical studies gave an insight into the effect of mgi mutations on the biochemistry of the ATP synthase, it is still not clear how the mutations convert a petite negative strain into a petite positive strain. There is a correlation between the percentage of petite mutations formed and the severity of the mutation on the coupling of the ATP synthase, but this is distinct from how uncoupling allows the petite negative cells to lose their mitochondrial DNA.
–
Mutations in the γ-subunit
The coiled-coil motif formed by the N- and C-terminal helices of the γ-subunit, rotates within the α3β3 complex. The rotation is believed to proceed with minimal resistance but maintains critical interactions to cause catalysis. Tethering the γ-subunit to the residues on either the α- or β-subunits inhibits rotation and blocks catalysis. When disulfide bridges were introduced in the E. coli ATPase between γ-Leu262Cys and α-Ala334Cys or between γ-Cys87 to β-Asp380Cys, the ATPase driven rotation was nearly completely inhibited [125]. However, rotation was not blocked when a covalent cross-link was made between γ-Ala285Cys and α-Pro280Cys. The location of the cross-links apparently provides an explanation to resolve this paradox. The cross links to γ-Leu262Cys and γ-Cys87 were positioned in the center or below the center of the rotor while the cross-link to γ-Ala285Cys was positioned at the top of the rotor. This study suggested that the amino acid residues at the top of the rotor could swivel along the c-α bonds or around the side chain bonds [126][127].
–
There are two isoforms of the γ-subunit in humans and other mammals: a “heart isoform” and a “liver isoform”. The isoforms differ by a single amino acid; the liver isoform has an additional residue, an aspartate, at the C-terminus. The isoforms are formed by alternative splicing, which is conserved and highly regulated [128][129][130][131]. The heart isoform is expressed in heart, skeletal muscle, and intercostal muscle, diaphragm, all containing tissues with a high and variable energy demand. The liver isoform is expressed in brain, thyroid, spleen, pancreas, kidney, testis and liver. Of these, all but the brain consume relatively low amount of ATP and have a steady energy demand [132]. The expectation is that these isoforms have biochemical significance and are important for the physiology of the tissues.
–
Based on the crystal structures of the bovine and yeast F1, we can predict that the Asp at the C-terminus of the γ-subunit in the liver isoform forms a salt linkage with α-Arg288. However, comparison of the ATPase activity of the two isoforms by both bulk ATPase activity measurements and analysis of rotation of single molecules did not demonstrate any significant difference in kinetics of ATPase reaction cycle [36][133]. This suggests that if the isoforms serve as a regulatory mechanism, that the regulation must occur in conjunction with another molecule.
–
Earlier studies on the bacterial enzyme have been critical in understanding the structure-function relationship of the γ-subunit of the ATP synthase. The “axle” of the bacterial and mitochondrial enzymes extends for about 22 residues on the N-terminus and for about 43 residues on the C-terminus (Figure 3). The helices formed by these regions form a coiled-coil that spins within the center of the α3β3 assembly. An early mutation isolated in E. coli γ-subunit gene, NR70, resulted in a 7 amino acid deletion, residues 22-28, in the bacterial γ-subunit [134][135]. The resultant bacteria were lacking membrane bound F1Fo and exhibited a reduced ability to accumulate sugars and amino acids but this could be restored by the addition DCCD, a covalent inhibitor of Fo. [134][135]. DCCD is apparently acting to prevent proton leakage through Fo. As such, this mutation exhibited qualities that are predicted for mutations that uncouple the ATP synthase, which allow free movement of protons.
–
A number of other studies have more closely defined the structure function relationship of the bacterial γ-subunit. Despite deletion of the axle region of the γ-subunit (the coiled-coil helices) the enzyme was still able to rotate in the correct direction with ATP hydrolysis. However, the rotation rate was greatly reduced and mutants with large deletions exhibited irregular motion [136]. Another study demonstrated that major deletions in the axle impaired ATP synthesis only by about 75% [137]. Based on the results from these and other studies, it was concluded that the C-terminal region of the γ-subunit is responsible for about half of the torque generated by ATP hydrolysis and the difference attributable to the interactions between the γ-subunit and residues at the orifice of the α3β3 subcomplex [138][139][140]. These results are also consistent with the location of the mgi mutations, which map at the top of the area surrounding the axle and at the general area of the orifice of the α3β3 subcomplex (Figure 3).
MUTATIONS IN THE ATP SYNTHASE THAT CAUSE HUMAN DISEASE
Disease-causing mutations in the nuclear DNA
There have been three reports of mutations in nuclear genes encoding subunits of the ATP synthase causing human disease: that encoding the ε-subunit [141] and more recently, the α-subunit [142][143]. In the first case, the mutation Tyr11 of the mature ε-subunit (Tyr12 in the precursor) was mutated to a Cys. Tyr11 is highly conserved in the ε-subunit in which the primary sequence is otherwise poorly conserved. The ε-subunit forms a two-helix hairpin and the N-terminus interacts with both the γ- and δ-subunits of the F1 ATPase. The side-chain phenolic group of Tyr11 is in the center of a pocket formed by residues (based on the structure of the bovine F1 ATPase, pdb: 1E79 [17]) Trp4, Ser10, Ile12, Arg13, Tyr14, and Ser15 of the ε-subunit, Val57, Leu58, Ser79 and Gly80 of the δ-subunit, and Asn203 and Glu206 of the γ-subunit. The mutation, ε-Tyr11Cys, resulted in a low level of the intact ATP synthase in the fibroblast cells of the patient, but there was an apparent associated increase in the level and activity of Complex III and IV of the electron transport chain. The ATP synthase that was assembled had reduced amount of associated ε-subunit, but nonetheless, was apparently functional. The patient was a 22 year old that presented with symptoms typical of a patient with a mitochondrial disease. This disease is not lethal and thus consistent with the impairment, but not elimination, of activity of the ATP synthase. The mutation likely alters the assembly of the intact ATP synthase.
–
The second example of a nuclear mutation in the ATP synthase causing a human disease was in the α-subunit, Tyr278Cys (Tyr321Cys of the pre-protein) [142]. In this case, the patient had symptoms that were typical of a severe mitochondrial disease resulting in death of the child at age 3. The patient had one sister with similar symptoms who died at 15 months. Both patients paradoxically had combined respiratory chain deficiency. Despite more severe symptoms and outcome, the Tyr278Cys mutation had a much less apparent effect on the function of the ATP synthase. The most pronounced effect of the mutations was observed in both the muscle and liver of the patient, where there was a moderate depletion of the mitochondrial DNA. A similar finding was observed when the comparable mutation was modeled in yeast [142]. Tyr278 is within the region where the mgi mutations were clustered in the yeast F1 ATPase suggesting that the mutation might affect the coupling capacity. The increased loss of mitochondrial DNA is also consistent with the mgi phenotype and thereby suggesting that the mutation might decrease the coupling of the ATP synthase. However, while there was some indication that the corresponding mutation when made in yeast uncoupled the mitochondrial ATP synthase, the effect was not large. One possible explanation for this paradox is that even mild uncoupling of the ATP synthase causes loss of mitochondrial DNA over many generations, acting analogous to a degenerative disease, and the loss in the mitochondrial DNA is ultimately the reason for the disease symptoms.
–
The third example was a disease presented in two siblings who contained a mutation in the α-subunit, Arg286Cys (Arg329 in the pre-protein) [143]. Both children died in the first weeks of life due to severe encephalopathy with associated extensive cerebral damage. In addition, there was damage to the lungs, kidney, and skeletal muscle. The mutation caused a dramatic decrease in the cellular level of the ATP synthase as measured by activity. The mutation was inherited from the father who was a heterozygote carrier. The mother did not have the effecting mutation but apparently had another mutation that caused loss of expression of the gene encoding the α-subunit from one of her homologs. Consistent with this, the level of mRNA for the α-subunit was decreased by 40% in the blood cells from the mother as compared to the father. As such, both patients were heterozygous for the Arg286Cys causing mutation and did not express any wild type form of the α-subunit. Complementation studies using cells derived from the patients indicated that the mutation in the gene encoding the α-subunit was responsible for the cellular defects. Unlike the mutation discussed earlier, this mutation did not have any apparent pleiotropic effect on the level of mitochondrial DNA or complexes of the electron transport chain.
–
These two reported disease-causing mutations in the gene encoding the α-subunit were recessive. Because there are three copies of the α-subunit in F1, assuming equal assembly between the mutant and wild type forms of the α-subunit, just 1/8th of the F1 would be assembled with all wild type α-subunit. As discussed earlier, a single mutation that inactivates the catalytic reaction will completely inhibit the enzyme activity [48]. As such, assuming equal probability of assembly of the wild type with the mutant forms, catalytic site mutations will inhibit activity by 7/8 of the total. Apparently, at least for these mutations, incorporation of a mutant copy of the α-subunit into the ATP synthase complex did not inactivate the catalytic site. Also, since the mother in the family with the Arg286Cys disease causing mutation expressed just 60% of the level of the α-subunit, a null mutation in the gene encoding the α-subunit causing a 50% decrease is unlikely to be a problem due to haploinsufficiency. Furthermore, there have been no reports to date on dominant mutations in the ATP synthase that are responsible for disease.
–
Disease-causing mutations in the mitochondrial DNA
The mitochondrial genome of humans encodes 2 subunits of the mitochondrial ATP synthase – subunits a and 8, with the remaining being encoded in the nucleus (c.f. Table 1). The majority of mutations in the ATP synthase that are associated with disease is located in the mitochondrial ATP6 gene, which encodes subunit-a. This is likely reflective of the importance of subunit-a in the proton translocation.
–
Disease-causing mutations in the genes encoded in the mitochondrial DNA were first identified in 1990 [144]. The mitochondrial genome is very compact, such that the open reading frames for subunits -a and -8 of the ATP synthase overlap with each other. To date, 29 disease- or phenotype-causing mutations have been reported in the ORF for subunit-a, subunit 6, and in the region that encodes both subunit-a and -8 (see www.mitomap.org for current listing).
–
Figure 4a shows a comparison of the primary sequence of subunit-a from E. coli, yeast, and human. Subunit-a is thought to be comprised of 5 transmembrane helices (TMH1-5) and the predicted TMH3-5 are as indicated. Figure 4b shows the possible arrangement of the transmembrane helices of subunits a (5 helices), b (2 helices) and c (20 helices) as determined by cross-linking and other biochemical studies [55][145][146][147][148][149]. The structure of subunit-a is not known, so this is just a hypothetical representation of the location of the putative helices. a-Arg186 in yeast (210 in E. coli, shaded blue) is conserved and thought to participate directly in the mechanism of proton pumping and thought to be closely associated with Glu59 in subunit-c. As such, in the model, the side chain of a-Arg186 is shown close to the carboxylate of c-Glu58. The residues shaded in yellow have been implicated to be associated with the proton half-channel (shown in the model as “On” and “Off” for the direction of proton movement during ATP synthesis) based on chemical modification studies using the enzyme isolated from E. coli [55][150]. Mutations in 8 residues in the human subunit, including a-Leu156, a-Leu217, and a-Leu220, are associated with diseases [144][151][152][153][154][155][156][157][158][159][160][161][162][163][164][165][166][167][168][169][170][171][172][173][174][175][176][177][178][179][180][181][182][183][184][185][186][187][188][189]. Residues a-Leu156 and a-Leu217 are highly conserved, adjacent to highly conserved, and located in TMH4 and TMH5 which contain clusters of residues that have been implicated in proton movement. a-Leu220 is adjacent to TMH5 and abuts highly conserved residues a-Tyr221 and a-Leu222. Based on the clustering of critical residues and the disease forming mutations, it seems likely that TMH4 and TMH5 are principle players in forming the proton half channels. An attractive hypothesis is that mutations in these residues alter proton movement indirectly by altering the secondary structure, for example a-Leu156Pro, a-Leu217Pro, and a-Leu222Pro or directly by impeding proton movement, as with a-Leu156Arg or a-Leu217Arg.
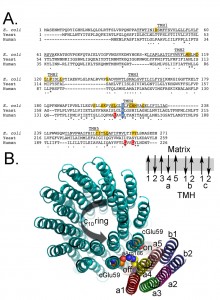 | FIGURE 4: Primary sequence alignment of subunit-a. (A) The partial primary sequence alignment of subunit-a from E. coli, yeast, and human is shown. The residues predicted to form transmembrane helices 3-5 (TMH3-5) are underlined and labeled [55][57][64][145]. The highly conserved and essential R210 (E. coli numbering) is shaded blue. The residues shaded red are mutated in the discussed human diseases. The residues shaded gold are identified as important for proton movement [55][190]. (B) Hypothetical model of the arrangement of the helices from subunits a, b, and c. The helices from subunits a (5 helices), b (2 helices) and c (20 helices) are shown along with the side chains for a-Arg1876 (yellow/blue) and c-Glu59 (red). There is very little cross-linking data positioning Helix 1 of subunit-a and thus the position is inferred from the absence, rather than presence, of cross-linking. Subunit b is thought to have a stoichiometry of 2 in the E. coli enzyme and 1 in the mitochondrial enzyme and there is 1 transmembrane helix in the bacterial subunit and 2 in the mitochondrial subunit. This view is from the matrix side of the mitochondrion. Also shown is the relative direction for deportation/protonation of c-Glu59 during ATP synthesis. The black arrow indicates the direction of rotation during ATP synthesis. Also shown is a schematic of the orientation of the transmembrane helices (TMH) for subunits a, b, and c in the membrane. Note that the helices for the subunits-a and -b are representations of a possible placement and secondary structure, while the structure of the c-ring is a crystal structure obtained from yeast [8]. |
The diseases associated with mutations in the gene encoding subunit-a vary in severity depending upon the allele, but the percentage of the mutant copy in each cell can vary greatly for 2 reasons. Mitochondrial DNA is typically a heteroplasmic mixture of mutant and wild type copies and further, mitochondrial DNA does not segregate in a Mendelian manner. For example, the mutations Leu156Arg, Leu156Pro, Leu217Arg, and Leu217Pro, manifest as NARP (Neurogenic muscle weakness, Ataxia, and Retinitis Pigmentosa) at low heteroplasmy but as Leigh syndrome (severe and fatal encephalopathy) when the percentage of mutant copies of mitochondrial DNA exceeds 90% (for review, see [191][192][193]). The disease phenotype is confounded by variations in the percentage of mitochondrial DNA molecules in the cells that contain the mutation, which can vary considerably between tissues and even between cells within a tissue – a situation referred to as “mosaic”. The varying genetic background of the patients, the tissue mosaicism, and the resulting variations in heteroplasmy can make interpretation of biochemical data and diagnosis difficult. To circumvent these problems, the effect of the mutation on the function of the ATP synthase has been evaluated by making the corresponding mutation in yeast or E. coli followed by biochemical analysis of the resulting mutant enzyme.
–
Using yeast as a model system, the effect of a deletion mutation in the gene encoding subunit-a on the assembly and function of the ATP synthase was assessed [194]. Subunit-a was shown to be necessary for the activity, but not for the assembly of the ATP synthase, and subcomplexes of
–
the enzyme can be formed devoid of subunit-a. The ATPase activity was at near normal levels but it was nearly insensitive to the Fo inhibitor, oligomycin. The cells rapidly lost mitochondrial DNA when the selection for the mitochondrial DNA was relaxed. The cells lost cytochrome oxidase activity, as was noted for deletion mutations in genes encoding other subunits of the ATP synthase discussed earlier. However, this study demonstrated that the loss of cytochrome oxidase activity was due to the loss in the synthesis of mitochondrial encoded subunit 1 of cytochrome oxidase, Cox1p. This causal relationship was further supported by studies in yeast and human fibroblast cells that showed that cytochrome oxidase activity was decreased in cells treated with oligomycin and partial recovery was caused by addition of an uncoupler [195]. In yeast, oligomycin caused a loss of Cox1p synthesis similar to that observed with the prior study with subunit-a deficient cells. These studies suggest that hyperpolarization of the mitochondrial membrane may compromise the synthesis of subunit 1 of cytochrome oxidase as addition of a limiting amount of uncoupler can restore cytochrome oxidase. Hyperpolarization of the mitochondrial membrane can be the result of mutation that inactivate or inhibit activity of the ATP synthase, thus establishing a possible link with cytochrome oxidase. Thus, as discussed earlier, there appears to be an evolutionary conserved link between a functional ATP synthase and synthesis of cytochrome oxidase and possibly the ΔΨ of the mitochondrial membrane is that link.
–
The biochemical consequences of a-Leu156Arg mutation were studied by making the corresponding mutation in E. coli [196]. The mutant enzyme had enzymatic properties that were nearly identical to the enzyme devoid of subunit-a. As such, in E. coli, mutations that affect the folding of subunit-a and assembly of the holo-enzyme cannot be firmly distinguished from mutations in subunit-a that do not prevent assembly, but rather impair the activity of the enzyme, simply by measuring the levels of ATP hydrolysis and synthesis. The analysis of the cultured fibroblasts [177] and cybrids [197] containing the Leu156Arg mutation indicated that the P/O ratio of the affected mitochondria [177] was not altered, but there was a decrease in both the level of ATP synthase and respiratory activity, and the mutation seemed to destabilize the complex [197]. Interestingly, the Leu156Arg mutation made the ATPase activity hypersensitive to oligomycin [177][197]. One explanation for this effect that is consistent with the mode of oligomycin binding to subunit c [53] is that the mutation in subunit-a made the binding site more accessible to oligomycin.
–
Similarly, subunit-a mutations, Leu217Arg, Leu217Pro, Leu156Arg, and Leu156Pro, were also studied using E. coli enzyme [161]. The mutation Leu217Arg did not largely alter the level of the ATP synthase, but severely inhibited ATP hydrolysis and ATP synthesis by the membrane bound enzyme. These results may indicate that the mutation blocks the proton conduction pathway in the ATP synthase without uncoupling the enzyme. In contrast, the Leu217Pro mutant form had considerable ATP synthase activity thus clearly showing biochemical differences due to the allelic differences. The mutations Leu156Arg and Leu156Pro caused diminished ATPase and ATP synthase activity but had little effect on the coupling of the enzyme. When the corresponding mutations, Leu156Pro and Leu156Arg, were made in yeast S. cerevisiae and analyzed, they demonstrated a decrease in the level of ATPase and ATP synthase activity, and again, a pleiotropic effect on the level of cytochrome oxidase activity [198][199]. For the yeast enzyme with Leu156Arg, the enzyme appeared to be fully assembled but defective in proton pumping and ATP synthesis. For the yeast enzyme with Leu156Pro, the decreased level of ATP synthase activity paralleled the decrease in oligomycin sensitivity suggesting that subunit-a was not efficiently incorporated into the ATP synthase. The effect of these mutations on the biochemistry of the bacterial and yeast enzymes were entirely consistent with the proposed role of subunit-a as providing a half-channel for proton access to the essential carboxylate in subunit-c, access to the intermembrane space, and the matrix space of the mitochondrion.
–
Disease-causing mutations in subunit 8 are much less common than those in subunit-a. The mutation Trp55X, where X is a stop codon, results in the truncation of 14 amino acids from the C-terminus of subunit 8 [200]. The resulting effect is a destabilization of the ATP synthase complex such that there was a large decrease in the level of intact ATP synthase complex and an increase in the amount of free F1 ATPase, as judged by Blue Native gels. It would be rather remarkable if the Blue Native gels accurately reflected the level of free F1 ATPase in the cells as a high level of free F1 would lead to a high level of futile and wasteful ATP hydrolysis. Possibly the ATP synthase complex is intact in the cell and only falls apart during the gel analysis. Alternatively, it is possible that the natural inhibitor protein, Inh1, is able to prevent the hydrolysis of ATP by free F1 ATPase. No other biochemical studies were done to give any insight into the effect of this mutation on the function of the ATP synthase.
–
There was a mutation in the overlapping region of the open reading frames of subunit-a and subunit-8 [201]. The mutation created a Thr55Arg replacement in subunit-8 and a Met1Thr replacement in subunit-a. The fibroblasts from the patient showed a significant, but not dramatic decrease in the ATP synthase activity driven by malate/glutamate or succinate. Likely, the change in the initiating codon for subunit-a resulted in a decrease in the level of subunit-a and thus in a decrease in the intact ATP synthase complex.
–
Single nucleotide polymorphisms (SNPs) in the nuclear genes encoding subunits of the ATP synthase
With the advent of rapid sequence analysis of the human genome, the bottleneck for the understanding of individual genetic differences is now the analysis of the effect of single nucleotide polymorphisms on biochemistry of the enzyme and the resulting effect on the physiology of the cell and organism. This analysis will be greatly aided by the understanding of the structure/function relationship of the protein and by the library of mutations that have already been studied. Nonetheless, the analysis is difficult and generally unpredictable – as was the case for the disease-causing mutation identified in the gene encoding the α-subunit and discussed earlier [142]. The difficulty can be illustrated by looking at the current library of the SNPs in the gene encoding the human γ-subunit of the ATP synthase (ATP5C1).
–
Many SNPs have been mapped in ATP5C1 (see http://evs.gs.washington.edu/EVS/). Note, mutations in genetic databases are normally numbered using the initiating Met as 1, while the convention used here and in structural and biochemical studies is that the first amino acid in the mature peptide is numbered 1. In the case of the human γ-subunit, there is a 25 amino acid leader polypeptide that targets the subunit to the mitochondria. Current SNPs that create missense mutations in the mature γ-subunit are: Ile6Val, Met25Thr, Ala32Val, Pro40Ser, Ala50Val, Ile85Val, Ile144Val, Ile169The, Ser186Asn, Tyr214Cys, Arg252Cys, Gly268Ser (Figure 5). γ-Met25 interacts with β-Ile390, which is in the Catch 2 region. The mutation in E. coli γ-subunit that corresponds to Met25Lys partially uncoupled the enzyme, reduced the binding affinity for Pi and reduced the apparent affinity for ATP of the E. coli enzyme [202][203][204]. However, it is not known if Met25Thr affects the biochemistry of the human enzyme.
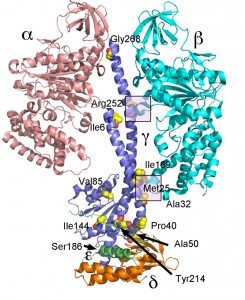 | FIGURE 5: Relative location in the ATP synthase of residues mutated by known single nucleotide polymorphisms. The structural representation is derived from the x-ray crystal structure of the bovine ATPase (1E79). To simplify the image, only a single copy of the α- (salmon) and β- (cyan) subunits is shown along with the γ- (purple), δ- (orange), and ε- (green) subunits. The residues, which are altered by known SNPs in the γ-subunit are shown in sphere representation and numbered using the human numbering system with 1 as the first residue in the mature peptide (the initiating Met is -25 for the γ-subunit). The two boxes represent the regions corresponding to Catch 1 and Catch 2 regions. |
The SNPs are not limited to the γ-subunit, but occur in some of the other genes encoding other subunits of the ATP synthase including the α- and β- subunits. Notwithstanding the putative importance of any one residue in the ATP synthase, the effect of a residue-altering mutation on the biochemistry of the enzyme is uncertain. Furthermore, the relationship between function of the ATP synthase and physiology of the organism is unclear. How much change in the coupling or activity of the ATP synthase is needed to have an observable effect on the individual? Do these SNPs account for differences in the individual’s performances in different climates, physical activities, or recovery from a trauma, such as a heart attack? The future lies in the ability to link SNPs with physical and medical risks or advantages for any one individual. Can we use yeast as a model organism to assess the effect of SNPs on the structure and activity of the enzyme?
–
A major impediment to the understanding of the ATP synthase is the lack of a high-resolution structure of the entire enzyme complex. The most pertinent portion of the enzyme that is lacking structural details is the proton half-channels, and this severely limits our understanding of the mechanism of proton translocation and pumping. The recent advances in electron microscopy make it inevitable that the structure of the ATP synthase will be solved but there is still hope that a high-resolution crystal structure will be obtained. However, structure determination of the mutant forms of the enzyme will likely also be necessary to fully understand the effect of disease causing mutations, especially in subunit a, and the effect of SNPs on the structure and function of the enzyme.
References
- P. MITCHELL, "Coupling of Phosphorylation to Electron and Hydrogen Transfer by a Chemi-Osmotic type of Mechanism", Nature, vol. 191, pp. 144-148, 1961. http://dx.doi.org/10.1038/191144a0
- P.D. Boyer, R.L. Cross, and W. Momsen, "A New Concept for Energy Coupling in Oxidative Phosphorylation Based on a Molecular Explanation of the Oxygen Exchange Reactions", Proceedings of the National Academy of Sciences, vol. 70, pp. 2837-2839, 1973. http://dx.doi.org/10.1073/pnas.70.10.2837
- I.N. Watt, M.G. Montgomery, M.J. Runswick, A.G.W. Leslie, and J.E. Walker, "Bioenergetic cost of making an adenosine triphosphate molecule in animal mitochondria", Proceedings of the National Academy of Sciences, vol. 107, pp. 16823-16827, 2010. http://dx.doi.org/10.1073/pnas.1011099107
- C.P. Lee, Q. Gu, Y. Xiong, R.A. Mitchell, and L. Ernster, "P/O ratios reassessed: mitochondrial P/O ratios consistently exceed 1.5 with succinate and 2.5 with NAD-linked substrates.", FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 1996. http://www.ncbi.nlm.nih.gov/pubmed/8641569
- P.C. Hinkle, "P/O ratios of mitochondrial oxidative phosphorylation", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1706, pp. 1-11, 2005. http://dx.doi.org/10.1016/j.bbabio.2004.09.004
- S.J. Kerscher, "Diversity and origin of alternative NADH:ubiquinone oxidoreductases", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1459, pp. 274-283, 2000. http://dx.doi.org/10.1016/S0005-2728(00)00162-6
- V. Kabaleeswaran, N. Puri, J.E. Walker, A.G.W. Leslie, and D.M. Mueller, "Novel features of the rotary catalytic mechanism revealed in the structure of yeast F1 ATPase", The EMBO Journal, vol. 25, pp. 5433-5442, 2006. http://dx.doi.org/10.1038/sj.emboj.7601410
- J. Symersky, V. Pagadala, D. Osowski, A. Krah, T. Meier, J.D. Faraldo-Gómez, and D.M. Mueller, "Structure of the c10 ring of the yeast mitochondrial ATP synthase in the open conformation", Nature Structural & Molecular Biology, vol. 19, pp. 485-491, 2012. http://dx.doi.org/10.1038/nsmb.2284
- D.M. Rees, A.G.W. Leslie, and J.E. Walker, "The structure of the membrane extrinsic region of bovine ATP synthase", Proceedings of the National Academy of Sciences, vol. 106, pp. 21597-21601, 2009. http://dx.doi.org/10.1073/pnas.0910365106
- V.K. Dickson, J.A. Silvester, I.M. Fearnley, A.G.W. Leslie, and J.E. Walker, "On the structure of the stator of the mitochondrial ATP synthase", The EMBO Journal, vol. 25, pp. 2911-2918, 2006. http://dx.doi.org/10.1038/sj.emboj.7601177
- J.E. Walker, and V.K. Dickson, "The peripheral stalk of the mitochondrial ATP synthase", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1757, pp. 286-296, 2006. http://dx.doi.org/10.1016/j.bbabio.2006.01.001
- J.P. Abrahams, A.G.W. Leslie, R. Lutter, and J.E. Walker, "Structure at 2.8 Â resolution of F1-ATPase from bovine heart mitochondria", Nature, vol. 370, pp. 621-628, 1994. http://dx.doi.org/10.1038/370621a0
- D.M. Rees, M.G. Montgomery, A.G.W. Leslie, and J.E. Walker, "Structural evidence of a new catalytic intermediate in the pathway of ATP hydrolysis by F 1 –ATPase from bovine heart mitochondria", Proceedings of the National Academy of Sciences, vol. 109, pp. 11139-11143, 2012. http://dx.doi.org/10.1073/pnas.1207587109
- J.P. Abrahams, S.K. Buchanan, M.J. Van Raaij, I.M. Fearnley, A.G. Leslie, and J.E. Walker, "The structure of bovine F1-ATPase complexed with the peptide antibiotic efrapeptin.", Proceedings of the National Academy of Sciences, vol. 93, pp. 9420-9424, 1996. http://dx.doi.org/10.1073/pnas.93.18.9420
- M.J. van Raaij, J.P. Abrahams, A.G. Leslie, and J.E. Walker, "The structure of bovine F1-ATPase complexed with the antibiotic inhibitor aurovertin B.", Proceedings of the National Academy of Sciences, vol. 93, pp. 6913-6917, 1996. http://dx.doi.org/10.1073/pnas.93.14.6913
- K. Braig, R.I. Menz, M.G. Montgomery, A.G. Leslie, and J.E. Walker, "Structure of bovine mitochondrial F1-ATPase inhibited by Mg2+ADP and aluminium fluoride", Structure, vol. 8, pp. 567-573, 2000. http://dx.doi.org/10.1016/S0969-2126(00)00145-3
- A.G.W. Leslie, J.E. Walker, C. Gibbons, and M.G. Montgomery, "Array", Nature Structural Biology, vol. 7, pp. 1055-1061, 2000. http://dx.doi.org/10.1038/80981
- R. Menz, J.E. Walker, and A.G. Leslie, "Structure of Bovine Mitochondrial F1-ATPase with Nucleotide Bound to All Three Catalytic Sites", Cell, vol. 106, pp. 331-341, 2001. http://dx.doi.org/10.1016/S0092-8674(01)00452-4
- R. Kagawa, M.G. Montgomery, K. Braig, A.G.W. Leslie, and J.E. Walker, "The structure of bovine F1-ATPase inhibited by ADP and beryllium fluoride", The EMBO Journal, vol. 23, pp. 2734-2744, 2004. http://dx.doi.org/10.1038/sj.emboj.7600293
- M.W. Bowler, M.G. Montgomery, A.G.W. Leslie, and J.E. Walker, "How azide inhibits ATP hydrolysis by the F-ATPases", Proceedings of the National Academy of Sciences, vol. 103, pp. 8646-8649, 2006. http://dx.doi.org/10.1073/pnas.0602915103
- M.W. Bowler, M.G. Montgomery, A.G. Leslie, and J.E. Walker, "Ground State Structure of F1-ATPase from Bovine Heart Mitochondria at 1.9 Aå Resolution", Journal of Biological Chemistry, vol. 282, pp. 14238-14242, 2007. http://dx.doi.org/10.1074/jbc.M700203200
- J.R. Gledhill, M.G. Montgomery, A.G.W. Leslie, and J.E. Walker, "Mechanism of inhibition of bovine F 1 -ATPase by resveratrol and related polyphenols", Proceedings of the National Academy of Sciences, vol. 104, pp. 13632-13637, 2007. http://dx.doi.org/10.1073/pnas.0706290104
- M.A. Bianchet, J. Hullihen, P.L. Pedersen, and L.M. Amzel, "The 2.8-Å structure of rat liver F 1 -ATPase: Configuration of a critical intermediate in ATP synthesis/hydrolysis", Proceedings of the National Academy of Sciences, vol. 95, pp. 11065-11070, 1998. http://dx.doi.org/10.1073/pnas.95.19.11065
- R.K. NAKAMOTO, K. SHIN, A. IWAMOTO, H. OMOTE, M. MAEDA, and M. FUTAI, "Escherichia coli F0F1‐ATPase", Annals of the New York Academy of Sciences, vol. 671, pp. 335-344, 1992. http://dx.doi.org/10.1111/j.1749-6632.1992.tb43807.x
- H. Begusch, and B. Hess, "Subunit dissociation—association of mitochondrial F1‐ATPase from yeast", FEBS Letters, vol. 108, pp. 249-252, 1979. http://dx.doi.org/10.1016/0014-5793(79)81221-1
- M. Dittrich, S. Hayashi, and K. Schulten, "On the Mechanism of ATP Hydrolysis in F1-ATPase", Biophysical Journal, vol. 85, pp. 2253-2266, 2003. http://dx.doi.org/10.1016/S0006-3495(03)74650-5
- Y. Komoriya, T. Ariga, R. Iino, H. Imamura, D. Okuno, and H. Noji, "Principal Role of the Arginine Finger in Rotary Catalysis of F1-ATPase", Journal of Biological Chemistry, vol. 287, pp. 15134-15142, 2012. http://dx.doi.org/10.1074/jbc.M111.328153
- I. Arnold, "Yeast mitochondrial F1F0-ATP synthase exists as a dimer: identification of three dimer-specific subunits", The EMBO Journal, vol. 17, pp. 7170-7178, 1998. http://dx.doi.org/10.1093/emboj/17.24.7170
- H. Noji, R. Yasuda, M. Yoshida, and K. Kinosita, "Direct observation of the rotation of F1-ATPase", Nature, vol. 386, pp. 299-302, 1997. http://dx.doi.org/10.1038/386299a0
- H. Itoh, A. Takahashi, K. Adachi, H. Noji, R. Yasuda, M. Yoshida, and K. Kinosita, "Mechanically driven ATP synthesis by F1-ATPase", Nature, vol. 427, pp. 465-468, 2004. http://dx.doi.org/10.1038/nature02212
- Y. Rondelez, G. Tresset, T. Nakashima, Y. Kato-Yamada, H. Fujita, S. Takeuchi, and H. Noji, "Highly coupled ATP synthesis by F1-ATPase single molecules", Nature, vol. 433, pp. 773-777, 2005. http://dx.doi.org/10.1038/nature03277
- R. Yasuda, H. Noji, M. Yoshida, K. Kinosita, and H. Itoh, "Resolution of distinct rotational substeps by submillisecond kinetic analysis of F1-ATPase", Nature, vol. 410, pp. 898-904, 2001. http://dx.doi.org/10.1038/35073513
- R. Yasuda, H. Noji, K. Kinosita, and M. Yoshida, "F1-ATPase Is a Highly Efficient Molecular Motor that Rotates with Discrete 120° Steps", Cell, vol. 93, pp. 1117-1124, 1998. http://dx.doi.org/10.1016/S0092-8674(00)81456-7
- T. Nishizaka, K. Oiwa, H. Noji, S. Kimura, E. Muneyuki, M. Yoshida, and K. Kinosita, "Chemomechanical coupling in F1-ATPase revealed by simultaneous observation of nucleotide kinetics and rotation", Nature Structural & Molecular Biology, vol. 11, pp. 142-148, 2004. http://dx.doi.org/10.1038/nsmb721
- K. Adachi, K. Oiwa, T. Nishizaka, S. Furuike, H. Noji, H. Itoh, M. Yoshida, and K. Kinosita, "Coupling of Rotation and Catalysis in F1-ATPase Revealed by Single-Molecule Imaging and Manipulation", Cell, vol. 130, pp. 309-321, 2007. http://dx.doi.org/10.1016/j.cell.2007.05.020
- B.C. Steel, A.L. Nord, Y. Wang, V. Pagadala, D.M. Mueller, and R.M. Berry, "Comparison between single-molecule and X-ray crystallography data on yeast F1-ATPase", Scientific Reports, vol. 5, 2015. http://dx.doi.org/10.1038/srep08773
- M. Nakanishi-Matsui, S. Kashiwagi, H. Hosokawa, D.J. Cipriano, S.D. Dunn, Y. Wada, and M. Futai, "Stochastic High-speed Rotation of Escherichia coli ATP Synthase F1 Sector", Journal of Biological Chemistry, vol. 281, pp. 4126-4131, 2006. http://dx.doi.org/10.1074/jbc.M510090200
- H. Omote, N. Sambonmatsu, K. Saito, Y. Sambongi, A. Iwamoto-Kihara, T. Yanagida, Y. Wada, and M. Futai, "The γ-subunit rotation and torque generation in F 1 -ATPase from wild-type or uncoupled mutant Escherichia coli", Proceedings of the National Academy of Sciences, vol. 96, pp. 7780-7784, 1999. http://dx.doi.org/10.1073/pnas.96.14.7780
- T. Bilyard, M. Nakanishi-Matsui, B.C. Steel, T. Pilizota, A.L. Nord, H. Hosokawa, M. Futai, and R.M. Berry, "High-resolution single-molecule characterization of the enzymatic states in Escherichia coli F 1 -ATPase", Philosophical Transactions of the Royal Society B: Biological Sciences, vol. 368, pp. 20120023, 2013. http://dx.doi.org/10.1098/rstb.2012.0023
- D. Spetzler, J. York, D. Daniel, R. Fromme, D. Lowry, and W. Frasch, "Microsecond Time Scale Rotation Measurements of Single F1-ATPase Molecules", Biochemistry, vol. 45, pp. 3117-3124, 2006. http://dx.doi.org/10.1021/bi052363n
- J.L. Martin, R. Ishmukhametov, T. Hornung, Z. Ahmad, and W.D. Frasch, "Anatomy of F 1 -ATPase powered rotation", Proceedings of the National Academy of Sciences, vol. 111, pp. 3715-3720, 2014. http://dx.doi.org/10.1073/pnas.1317784111
- T. Suzuki, K. Tanaka, C. Wakabayashi, E. Saita, and M. Yoshida, "Chemomechanical coupling of human mitochondrial F1-ATPase motor", Nature Chemical Biology, vol. 10, pp. 930-936, 2014. http://dx.doi.org/10.1038/nchembio.1635
- G.L. Choate, R.L. Hutton, and P.D. Boyer, "Occurrence and significance of oxygen exchange reactions catalyzed by mitochondrial adenosine triphosphatase preparations.", The Journal of biological chemistry, 1979. http://www.ncbi.nlm.nih.gov/pubmed/153910
- C. Grubmeyer, R.L. Cross, and H.S. Penefsky, "Mechanism of ATP hydrolysis by beef heart mitochondrial ATPase. Rate constants for elementary steps in catalysis at a single site.", The Journal of biological chemistry, 1982. http://www.ncbi.nlm.nih.gov/pubmed/6214557
- R.L. Cross, C. Grubmeyer, and H.S. Penefsky, "Mechanism of ATP hydrolysis by beef heart mitochondrial ATPase. Rate enhancements resulting from cooperative interactions between multiple catalytic sites.", The Journal of biological chemistry, 1982. http://www.ncbi.nlm.nih.gov/pubmed/6214558
- D.M. MUELLER, V. INDYK, and L. MCGILL, "ATPase kinetics for wild‐type Saccharomyces cerevisiae F1‐ATPase and F1‐ATPase with the β‐subunit Thr197→Ser mutation", European Journal of Biochemistry, vol. 222, pp. 991-999, 1994. http://dx.doi.org/10.1111/j.1432-1033.1994.tb18950.x
- K. Kinosita, K. Adachi, and H. Itoh, "Rotation of F1-ATPase: How an ATP-Driven Molecular Machine May Work", Annual Review of Biophysics and Biomolecular Structure, vol. 33, pp. 245-268, 2004. http://dx.doi.org/10.1146/annurev.biophys.33.110502.132716
- T. Amano, T. Hisabori, E. Muneyuki, and M. Yoshida, "Catalytic Activities of α3β3γ Complexes of F1-ATPase with 1, 2, or 3 Incompetent Catalytic Sites", Journal of Biological Chemistry, vol. 271, pp. 18128-18133, 1996. http://dx.doi.org/10.1074/jbc.271.30.18128
- T. Ariga, T. Masaike, H. Noji, and M. Yoshida, "Stepping Rotation of F1-ATPase with One, Two, or Three Altered Catalytic Sites That Bind ATP Only Slowly", Journal of Biological Chemistry, vol. 277, pp. 24870-24874, 2002. http://dx.doi.org/10.1074/jbc.M202582200
- Y. Kagawa, and E. Racker, "Partial resolution of the enzymes catalyzing oxidative phosphorylation. 8. Properties of a factor conferring oligomycin sensitivity on mitochondrial adenosine triphosphatase.", The Journal of biological chemistry, 1966. http://www.ncbi.nlm.nih.gov/pubmed/4223640
- E. Racker, "A reconstituted system of oxidative phosphorylation", Biochemical and Biophysical Research Communications, vol. 14, pp. 75-78, 1963. http://dx.doi.org/10.1016/0006-291x(63)90214-6
- E. Racker, "A mitochondrial factor conferring oligomycin sensitivity on soluble mitochondrial ATPase", Biochemical and Biophysical Research Communications, vol. 10, pp. 435-439, 1963. http://dx.doi.org/10.1016/0006-291x(63)90375-9
- J. Symersky, D. Osowski, D.E. Walters, and D.M. Mueller, "Oligomycin frames a common drug-binding site in the ATP synthase", Proceedings of the National Academy of Sciences, vol. 109, pp. 13961-13965, 2012. http://dx.doi.org/10.1073/pnas.1207912109
- I.R. Collinson, M.J. Runswick, S.K. Buchanan, I.M. Fearnley, J.M. Skehel, M.J. van Raaij, D.E. Griffiths, and J.E. Walker, "F0 Membrane Domain of ATP Synthase from Bovine Heart Mitochondria: Purification, Subunit Composition, and Reconstitution with F1-ATPase", Biochemistry, vol. 33, pp. 7971-7978, 1994. http://dx.doi.org/10.1021/bi00191a026
- H. Dong, and R.H. Fillingame, "Chemical Reactivities of Cysteine Substitutions in Subunit a of ATP Synthase Define Residues Gating H+ Transport from Each Side of the Membrane", Journal of Biological Chemistry, vol. 285, pp. 39811-39818, 2010. http://dx.doi.org/10.1074/jbc.M110.175844
- R.H. Fillingame, C.M. Angevine, and O.Y. Dmitriev, "Mechanics of coupling proton movements to c‐ring rotation in ATP synthase", FEBS Letters, vol. 555, pp. 29-34, 2003. http://dx.doi.org/10.1016/S0014-5793(03)01101-3
- J.C. Long, S. Wang, and S.B. Vik, "Membrane Topology of Subunit a of the F1F0 ATP Synthase as Determined by Labeling of Unique Cysteine Residues", Journal of Biological Chemistry, vol. 273, pp. 16235-16240, 1998. http://dx.doi.org/10.1074/jbc.273.26.16235
- L.A. Baker, I.N. Watt, M.J. Runswick, J.E. Walker, and J.L. Rubinstein, "Arrangement of subunits in intact mammalian mitochondrial ATP synthase determined by cryo-EM", Proceedings of the National Academy of Sciences, vol. 109, pp. 11675-11680, 2012. http://dx.doi.org/10.1073/pnas.1204935109
- D. Stock, A.G.W. Leslie, and J.E. Walker, "Molecular Architecture of the Rotary Motor in ATP Synthase", Science, vol. 286, pp. 1700-1705, 1999. http://dx.doi.org/10.1126/science.286.5445.1700
- T. Meier, P. Polzer, K. Diederichs, W. Welte, and P. Dimroth, "Structure of the Rotor Ring of F-Type Na + -ATPase from Ilyobacter tartaricus", Science, vol. 308, pp. 659-662, 2005. http://dx.doi.org/10.1126/science.1111199
- D. Matthies, L. Preiss, A.L. Klyszejko, D.J. Muller, G.M. Cook, J. Vonck, and T. Meier, "The c13 Ring from a Thermoalkaliphilic ATP Synthase Reveals an Extended Diameter Due to a Special Structural Region", Journal of Molecular Biology, vol. 388, pp. 611-618, 2009. http://dx.doi.org/10.1016/j.jmb.2009.03.052
- M. Vollmar, D. Schlieper, M. Winn, C. Büchner, and G. Groth, "Structure of the c14 Rotor Ring of the Proton Translocating Chloroplast ATP Synthase", Journal of Biological Chemistry, vol. 284, pp. 18228-18235, 2009. http://dx.doi.org/10.1074/jbc.M109.006916
- D. Pogoryelov, �. Yildiz, J.D. Faraldo-Gómez, and T. Meier, "High-resolution structure of the rotor ring of a proton-dependent ATP synthase", Nature Structural & Molecular Biology, vol. 16, pp. 1068-1073, 2009. http://dx.doi.org/10.1038/nsmb.1678
- S.B. Vik, J.C. Long, T. Wada, and D. Zhang, "A model for the structure of subunit a of the Escherichia coli ATP synthase and its role in proton translocation", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1458, pp. 457-466, 2000. http://dx.doi.org/10.1016/s0005-2728(00)00094-3
- W.C.Y. Lau, and J.L. Rubinstein, "Structure of intact Thermus thermophilus V-ATPase by cryo-EM reveals organization of the membrane-bound V O motor", Proceedings of the National Academy of Sciences, vol. 107, pp. 1367-1372, 2010. http://dx.doi.org/10.1073/pnas.0911085107
- J. Velours, J. Vaillier, P. Paumard, V. Soubannier, J. Lai-Zhang, and D.M. Mueller, "Bovine Coupling Factor 6, with Just 14.5% Shared Identity, Replaces Subunit h in the Yeast ATP Synthase", Journal of Biological Chemistry, vol. 276, pp. 8602-8607, 2001. http://dx.doi.org/10.1074/jbc.m008123200
- J.E. Walker, R. Lutter, A. Dupuis, and M.J. Runswick, "Identification of the subunits of F1F0-ATPase from bovine heart mitochondria", Biochemistry, vol. 30, pp. 5369-5378, 1991. http://dx.doi.org/10.1021/bi00236a007
- C. Spannagel, J. Vaillier, G. Arselin, P. Graves, and J. Velours, "The Subunit F of Mitochondrial Yeast ATP Synthase", European Journal of Biochemistry, vol. 247, pp. 1111-1117, 1997. http://dx.doi.org/10.1111/j.1432-1033.1997.01111.x
- J. Vaillier, G. Arselin, P. Graves, N. Camougrand, and J. Velours, "Isolation of Supernumerary Yeast ATP Synthase Subunits e and i", Journal of Biological Chemistry, vol. 274, pp. 543-548, 1999. http://dx.doi.org/10.1074/jbc.274.1.543
- I. Arnold, K. Pfeiffer, W. Neupert, R.A. Stuart, and H. Schägger, "ATP Synthase of Yeast Mitochondria", Journal of Biological Chemistry, vol. 274, pp. 36-40, 1999. http://dx.doi.org/10.1074/jbc.274.1.36
- J. Lai-Zhang, "Epistatic interactions of deletion mutants in the genes encoding the F1-ATPase in yeast Saccharomyces cerevisiae", The EMBO Journal, vol. 18, pp. 58-64, 1999. http://dx.doi.org/10.1093/emboj/18.1.58
- K.M. Davies, M. Strauss, B. Daum, J.H. Kief, H.D. Osiewacz, A. Rycovska, V. Zickermann, and W. Kühlbrandt, "Macromolecular organization of ATP synthase and complex I in whole mitochondria", Proceedings of the National Academy of Sciences, vol. 108, pp. 14121-14126, 2011. http://dx.doi.org/10.1073/pnas.1103621108
- H. Seelert, and N.A. Dencher, "ATP synthase superassemblies in animals and plants: Two or more are better", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1807, pp. 1185-1197, 2011. http://dx.doi.org/10.1016/j.bbabio.2011.05.023
- P. Paumard, J. Vaillier, B. Coulary, J. Schaeffer, V. Soubannier, D.M. Mueller, D. Brèthes, J. di Rago, and J. Velours, "The ATP synthase is involved in generating mitochondrial cristae morphology", The EMBO Journal, vol. 21, pp. 221-230, 2002. http://dx.doi.org/10.1093/emboj/21.3.221
- G. Arselin, J. Vaillier, B. Salin, J. Schaeffer, M. Giraud, A. Dautant, D. Brèthes, and J. Velours, "The Modulation in Subunits e and g Amounts of Yeast ATP Synthase Modifies Mitochondrial Cristae Morphology", Journal of Biological Chemistry, vol. 279, pp. 40392-40399, 2004. http://dx.doi.org/10.1074/jbc.m404316200
- G.M. Boyle, X. Roucou, P. Nagley, R.J. Devenish, and M. Prescott, "Identification of subunit g of yeast mitochondrial F1F0‐ATP synthase, a protein required for maximal activity of cytochrome c oxidase", European Journal of Biochemistry, vol. 262, pp. 315-323, 1999. http://dx.doi.org/10.1046/j.1432-1327.1999.00345.x
- S. Saddar, M.K. Dienhart, and R.A. Stuart, "The F1F0-ATP Synthase Complex Influences the Assembly State of the Cytochrome bc1-Cytochrome Oxidase Supercomplex and Its Association with the TIM23 Machinery", Journal of Biological Chemistry, vol. 283, pp. 6677-6686, 2008. http://dx.doi.org/10.1074/jbc.M708440200
- H. Shen, D.E. Walters, and D.M. Mueller, "Introduction of the Chloroplast Redox Regulatory Region in the Yeast ATP Synthase Impairs Cytochrome c Oxidase", Journal of Biological Chemistry, vol. 283, pp. 32937-32943, 2008. http://dx.doi.org/10.1074/jbc.M805310200
- M.E. PULLMAN, and G.C. MONROY, "A NATURALLY OCCURRING INHIBITOR OF MITOCHONDRIAL ADENOSINE TRIPHOSPHATASE.", The Journal of biological chemistry, 1963. http://www.ncbi.nlm.nih.gov/pubmed/14109217
- A. Akashi, Y. Yoshida, H. Nakagoshi, K. Kuroki, T. Hashimoto, K. Tagawa, and F. Imamoto, "Molecular cloning and expression of a gene for a factor which stabilizes formation of inhibitor-mitochondrial ATPase complex from Saccharomyces cerevisiae.", Journal of biochemistry, 1988. http://www.ncbi.nlm.nih.gov/pubmed/2907329
- N. Ichikawa, Y. Yoshida, T. Hashimoto, N. Ogasawara, H. Yoshikawa, F. Imamoto, and K. Tagawa, "Activation of ATP hydrolysis by an uncoupler in mutant mitochondria lacking an intrinsic ATPase inhibitor in yeast.", The Journal of biological chemistry, 1990. http://www.ncbi.nlm.nih.gov/pubmed/2138617
- W. Rouslin, and C.W. Broge, "Mechanisms of ATP conservation during ischemia in slow and fast heart rate hearts.", The American journal of physiology, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8430769
- W. Rouslin, "Regulation of the mitochondrial ATPasein situ in cardiac muscle: Role of the inhibitor subunit", Journal of Bioenergetics and Biomembranes, vol. 23, pp. 873-888, 1991. http://dx.doi.org/10.1007/bf00786006
- W. Rouslin, and C.W. Broge, "Regulation of mitochondrial matrix pH and adenosine 5'-triphosphatase activity during ischemia in slow heart-rate hearts. Role of Pi/H+ symport.", The Journal of biological chemistry, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2527849
- W. Rouslin, "Factors affecting the reactivation of the oligomycin-sensitive adenosine 5'-triphosphatase and the release of ATPase inhibitor protein during the re-energization of intact mitochondria from ischemic cardiac muscle.", The Journal of biological chemistry, 1987. http://www.ncbi.nlm.nih.gov/pubmed/2950098
- W. ROUSLIN, and J. ERICKSON, "Factors affecting the loss of mitochondrial function in autolyzing cardiac muscle", Journal of Molecular and Cellular Cardiology, vol. 18, pp. 1187-1195, 1986. http://dx.doi.org/10.1016/s0022-2828(86)80044-x
- M. Campanella, E. Casswell, S. Chong, Z. Farah, M.R. Wieckowski, A.Y. Abramov, A. Tinker, and M.R. Duchen, "Regulation of Mitochondrial Structure and Function by the F1Fo-ATPase Inhibitor Protein, IF1", Cell Metabolism, vol. 8, pp. 13-25, 2008. http://dx.doi.org/10.1016/j.cmet.2008.06.001
- M. Rak, S. Gokova, and A. Tzagoloff, "Modular assembly of yeast mitochondrial ATP synthase", The EMBO Journal, vol. 30, pp. 920-930, 2011. http://dx.doi.org/10.1038/emboj.2010.364
- G. López-Martínez, B. Rodríguez-Porrata, M. Margalef-Català, and R. Cordero-Otero, "The STF2p Hydrophilin from Saccharomyces cerevisiae Is Required for Dehydration Stress Tolerance", PLoS ONE, vol. 7, pp. e33324, 2012. http://dx.doi.org/10.1371/journal.pone.0033324
- M. Fujikawa, H. Imamura, J. Nakamura, and M. Yoshida, "Assessing Actual Contribution of IF1, Inhibitor of Mitochondrial FoF1, to ATP Homeostasis, Cell Growth, Mitochondrial Morphology, and Cell Viability", Journal of Biological Chemistry, vol. 287, pp. 18781-18787, 2012. http://dx.doi.org/10.1074/jbc.M112.345793
- E. Cabezón, M.G. Montgomery, A.G.W. Leslie, and J.E. Walker, "The structure of bovine F1-ATPase in complex with its regulatory protein IF1", Nature Structural & Molecular Biology, vol. 10, pp. 744-750, 2003. http://dx.doi.org/10.1038/nsb966
- J.V. Bason, M.G. Montgomery, A.G.W. Leslie, and J.E. Walker, "Pathway of binding of the intrinsically disordered mitochondrial inhibitor protein to F 1 -ATPase", Proceedings of the National Academy of Sciences, vol. 111, pp. 11305-11310, 2014. http://dx.doi.org/10.1073/pnas.1411560111
- S.P. Tsunoda, A.J.W. Rodgers, R. Aggeler, M.C.J. Wilce, M. Yoshida, and R.A. Capaldi, "Large conformational changes of the ɛ subunit in the bacterial F 1 F 0 ATP synthase provide a ratchet action to regulate this rotary motor enzyme", Proceedings of the National Academy of Sciences, vol. 98, pp. 6560-6564, 2001. http://dx.doi.org/10.1073/pnas.111128098
- J.M. Rizzo, M. Tarsio, G.A. Martínez-Muñoz, and P.M. Kane, "Diploids Heterozygous for a vma13Δ Mutation in Saccharomyces cerevisiae Highlight the Importance of V-ATPase Subunit Balance in Supporting Vacuolar Acidification and Silencing Cytosolic V1-ATPase Activity", Journal of Biological Chemistry, vol. 282, pp. 8521-8532, 2007. http://dx.doi.org/10.1074/jbc.M607092200
- S.C. Li, T.T. Diakov, J.M. Rizzo, and P.M. Kane, "Vacuolar H + -ATPase Works in Parallel with the HOG Pathway To Adapt Saccharomyces cerevisiae Cells to Osmotic Stress", Eukaryotic Cell, vol. 11, pp. 282-291, 2012. http://dx.doi.org/10.1128/EC.05198-11
- S.H. Ackerman, "Atp11p and Atp12p are chaperones for F1-ATPase biogenesis in mitochondria", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1555, pp. 101-105, 2002. http://dx.doi.org/10.1016/s0005-2728(02)00262-1
- Z. Wang, P.S. White, and S.H. Ackerman, "Atp11p and Atp12p Are Assembly Factors for the F1-ATPase in Human Mitochondria", Journal of Biological Chemistry, vol. 276, pp. 30773-30778, 2001. http://dx.doi.org/10.1074/jbc.m104133200
- Z. Wang, and S.H. Ackerman, "The Assembly Factor Atp11p Binds to the β-Subunit of the Mitochondrial F1-ATPase", Journal of Biological Chemistry, vol. 275, pp. 5767-5772, 2000. http://dx.doi.org/10.1074/jbc.275.8.5767
- Z. Wang, and S.H. Ackerman, "Identification of Functional Domains in Atp11p", Journal of Biological Chemistry, vol. 271, pp. 4887-4894, 1996. http://dx.doi.org/10.1074/jbc.271.9.4887
- S.H. Ackerman, and A. Tzagoloff, "Identification of two nuclear genes (ATP11, ATP12) required for assembly of the yeast F1-ATPase.", Proceedings of the National Academy of Sciences, vol. 87, pp. 4986-4990, 1990. http://dx.doi.org/10.1073/pnas.87.13.4986
- Z. Wang, and S.H. Ackerman, "Mutational Studies with Atp12p, a Protein Required for Assembly of the Mitochondrial F1-ATPase in Yeast", Journal of Biological Chemistry, vol. 273, pp. 2993-3002, 1998. http://dx.doi.org/10.1074/jbc.273.5.2993
- Y. Xiao, M. Metzl, and D.M. Mueller, "Partial Uncoupling of the Mitochondrial Membrane by a Heterozygous Null Mutation in the Gene Encoding the γ- or δ-Subunit of the Yeast Mitochondrial ATPase", Journal of Biological Chemistry, vol. 275, pp. 6963-6968, 2000. http://dx.doi.org/10.1074/jbc.275.10.6963
- S. Duvezin-Caubet, M. Caron, M. Giraud, J. Velours, and J. di Rago, "The two rotor components of yeast mitochondrial ATP synthase are mechanically coupled by subunit δ", Proceedings of the National Academy of Sciences, vol. 100, pp. 13235-13240, 2003. http://dx.doi.org/10.1073/pnas.2135169100
- D.M. Mueller, "Array", Journal of Bioenergetics and Biomembranes, vol. 32, pp. 391-400, 2000. http://dx.doi.org/10.1023/A:1005532104617
- E.M. Beasley, C. Wachter, and G. Schatz, "Putting energy into mitochondrial protein import", Current Biology, vol. 2, pp. 520, 1992. http://dx.doi.org/10.1016/0960-9822(92)90004-t
- S.M. Gasser, G. Daum, and G. Schatz, "Import of proteins into mitochondria. Energy-dependent uptake of precursors by isolated mitochondria.", The Journal of biological chemistry, 1982. http://www.ncbi.nlm.nih.gov/pubmed/6215405
- M. KLINGENBERG, and H. ROTTENBERG, "Relation between the Gradient of the ATP/ADP Ratio and the Membrane Potential across the Mitochondrial Membrane", European Journal of Biochemistry, vol. 73, pp. 125-130, 1977. http://dx.doi.org/10.1111/j.1432-1033.1977.tb11298.x
- C. Dupont, J.P. Mazat, and B. Guerin, "The role of adenine nucleotide translocation in the energization of the inner membrane of mitochondria isolated from ϱ+ and ϱo strains of saccharomyces cerevisiae", Biochemical and Biophysical Research Communications, vol. 132, pp. 1116-1123, 1985. http://dx.doi.org/10.1016/0006-291X(85)91922-9
- D.J. Kominsky, M.P. Brownson, D.L. Updike, and P.E. Thorsness, "Genetic and biochemical basis for viability of yeast lacking mitochondrial genomes.", Genetics, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12524335
- J. Lai‐Zhang, and D.M. Mueller, "Complementation of deletion mutants in the genes encoding the F1‐ATPase by expression of the corresponding bovine subunits in yeast S. cerevisiae", European Journal of Biochemistry, vol. 267, pp. 2409-2418, 2000. http://dx.doi.org/10.1046/j.1432-1327.2000.01253.x
- E. Guélin, J. Chevallier, M. Rigoulet, B. Guérin, and J. Velours, "ATP synthase of yeast mitochondria. Isolation and disruption of the ATP epsilon gene.", The Journal of biological chemistry, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8416924
- E. Tetaud, F. Godard, M. Giraud, S.H. Ackerman, and J. di Rago, "The depletion of F1subunit ε in yeast leads to an uncoupled respiratory phenotype that is rescued by mutations in the proton-translocating subunits of F0", Molecular Biology of the Cell, vol. 25, pp. 791-799, 2014. http://dx.doi.org/10.1091/mbc.E13-02-0112
- G.D. Clark-Walker, and X.J. Chen, "A vital function for mitochondrial DNA in the petite-negative yeastKluyveromyces lactis", Molecular and General Genetics MGG, vol. 252, pp. 746-750, 1996. http://dx.doi.org/10.1007/BF02173982
- X.J. Chen, P.M. Hansbro, and G.D. Clark-Walker, "Suppression of ρ 0 lethality by mitochondrial ATP synthase F1 mutations in Kluyveromyces lactis occurs in the absence of F0", Molecular and General Genetics MGG, vol. 259, pp. 457-467, 1998. http://dx.doi.org/10.1007/s004380050836
- P.M. Hansbro, X.J. Chen, and G.D. Clark-Walker, "Allele-specific expression of the Mgi- phenotype on disruption of the F 1 -ATPase delta-subunit gene in Kluyveromyces lactis", Current Genetics, vol. 33, pp. 46-51, 1998. http://dx.doi.org/10.1007/s002940050307
- X.J. Chen, and G.D. Clark-Walker, "α and β subunits of F1-ATPase are required for survival of petite mutants in Saccharomyces cerevisiae", Molecular and General Genetics MGG, vol. 262, pp. 898-908, 1999. http://dx.doi.org/10.1007/s004380051156
- G. Clark-Walker, P.M. Hansbro, F. Gibson, and X.J. Chen, "Mutant residues suppressing ρ0-lethality in Kluyveromyces lactis occur at contact sites between subunits of F1-ATPase", Biochimica et Biophysica Acta (BBA) - Protein Structure and Molecular Enzymology, vol. 1478, pp. 125-137, 2000. http://dx.doi.org/10.1016/S0167-4838(00)00003-0
- C. Fernet, D. Clark-Walker, and M. Claisse, "The mitochondrial genome can be altered or lost without lethal effect in the petite-negative yeast Debaryomyces ( Schwanniomyces ) occidentalis", Current Genetics, vol. 42, pp. 94-102, 2002. http://dx.doi.org/10.1007/s00294-002-0337-4
- D.J. Kominsky, and P.E. Thorsness, "Expression of the Saccharomyces cerevisiae gene YME1 in the petite-negative yeast Schizosaccharomyces pombe converts it to petite-positive.", Genetics, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10628976
- E.R. Weber, R.S. Rooks, K.S. Shafer, J.W. Chase, and P.E. Thorsness, "Mutations in the mitochondrial ATP synthase gamma subunit suppress a slow-growth phenotype of yme1 yeast lacking mitochondrial DNA.", Genetics, 1995. http://www.ncbi.nlm.nih.gov/pubmed/7498726
- A. Schnaufer, G.D. Clark-Walker, A.G. Steinberg, and K. Stuart, "The F1-ATP synthase complex in bloodstream stage trypanosomes has an unusual and essential function", The EMBO Journal, vol. 24, pp. 4029-4040, 2005. http://dx.doi.org/10.1038/sj.emboj.7600862
- S. Dean, M.K. Gould, C.E. Dewar, and A.C. Schnaufer, "Single point mutations in ATP synthase compensate for mitochondrial genome loss in trypanosomes", Proceedings of the National Academy of Sciences, vol. 110, pp. 14741-14746, 2013. http://dx.doi.org/10.1073/pnas.1305404110
- Y. Wang, U. Singh, and D.M. Mueller, "Mitochondrial Genome Integrity Mutations Uncouple the Yeast Saccharomyces cerevisiae ATP Synthase", Journal of Biological Chemistry, vol. 282, pp. 8228-8236, 2007. http://dx.doi.org/10.1074/jbc.M609635200
- D. Arsenieva, J. Symersky, Y. Wang, V. Pagadala, and D.M. Mueller, "Crystal Structures of Mutant Forms of the Yeast F1 ATPase Reveal Two Modes of Uncoupling", Journal of Biological Chemistry, vol. 285, pp. 36561-36569, 2010. http://dx.doi.org/10.1074/jbc.M110.174383
- K. Gumbiowski, D. Cherepanov, M. Müller, O. Pänke, P. Promto, S. Winkler, W. Junge, and S. Engelbrecht, "F-ATPase: Forced Full Rotation of the Rotor Despite Covalent Cross-link with the Stator", Journal of Biological Chemistry, vol. 276, pp. 42287-42292, 2001. http://dx.doi.org/10.1074/jbc.M106884200
- M. Müller, K. Gumbiowski, D.A. Cherepanov, S. Winkler, W. Junge, S. Engelbrecht, and O. Pänke, "Rotary F1‐ATPase", European Journal of Biochemistry, vol. 271, pp. 3914-3922, 2004. http://dx.doi.org/10.1111/j.1432-1033.2004.04328.x
- F. Hilbers, W. Junge, and H. Sielaff, "The Torque of Rotary F-ATPase Can Unfold Subunit Gamma If Rotor and Stator Are Cross-Linked", PLoS ONE, vol. 8, pp. e53754, 2013. http://dx.doi.org/10.1371/journal.pone.0053754
- M. Ichida, H. Endo, U. Ikeda, C. Matsuda, E. Ueno, K. Shimada, and Y. Kagawa, "MyoD Is Indispensable for Muscle-specific Alternative Splicing in Mouse Mitochondrial ATP Synthase γ-Subunit Pre-mRNA", Journal of Biological Chemistry, vol. 273, pp. 8492-8501, 1998. http://dx.doi.org/10.1074/jbc.273.14.8492
- H. Endo, C. Matsuda, and Y. Kagawa, "Exclusion of an alternatively spliced exon in human ATP synthase gamma-subunit pre-mRNA requires de novo protein synthesis.", The Journal of biological chemistry, 1994. http://www.ncbi.nlm.nih.gov/pubmed/8175656
- M. Hayakawa, H. Endo, T. Hamamoto, and Y. Kagawa, "Acidic Stimulation Induces a Negative Regulatory Factor That Affects Alternative Exon Selectionin Vitroin Human ATP Synthase γ-Subunit Pre-mRNA", Biochemical and Biophysical Research Communications, vol. 251, pp. 603-608, 1998. http://dx.doi.org/10.1006/bbrc.1998.9525
- M. Hayakawa, E. Sakashita, E. Ueno, S. Tominaga, T. Hamamoto, Y. Kagawa, and H. Endo, "Muscle-specific Exonic Splicing Silencer for Exon Exclusion in Human ATP Synthase γ-Subunit Pre-mRNA", Journal of Biological Chemistry, vol. 277, pp. 6974-6984, 2002. http://dx.doi.org/10.1074/jbc.M110138200
- C. Matsuda, H. Endo, H. Hirata, H. Morosawa, M. Nakanishi, and Y. Kagawa, "Tissue‐specific isoforms of the bovine mitochondrial ATP synthase γ‐subunit", FEBS Letters, vol. 325, pp. 281-284, 1993. http://dx.doi.org/10.1016/0014-5793(93)81089-I
- C. Matsuda, E. Muneyuki, H. Endo, M. Yoshida, and Y. Kagawa, "Comparison of the ATPase Activities of Bovine Heart and Liver Mitochondrial ATP Synthases with Different Tissue-Specific γSubunit Isoforms", Biochemical and Biophysical Research Communications, vol. 200, pp. 671-678, 1994. http://dx.doi.org/10.1006/bbrc.1994.1503
- B.P. Rosen, "Restoration of active transport in an Mg2+-adenosine triphosphatase-deficient mutant of Escherichia coli.", Journal of bacteriology, 1973. http://www.ncbi.nlm.nih.gov/pubmed/4270946
- H. Kanazawa, H. Hama, B.P. Rosen, and M. Futai, "Deletion of seven amino acid residues from the γ subunit of Escherichia coli H+-ATPase causes total loss of F1 assembly on membranes", Archives of Biochemistry and Biophysics, vol. 241, pp. 364-370, 1985. http://dx.doi.org/10.1016/0003-9861(85)90558-2
- S. Furuike, M.D. Hossain, Y. Maki, K. Adachi, T. Suzuki, A. Kohori, H. Itoh, M. Yoshida, and K. Kinosita, "Axle-Less F 1 -ATPase Rotates in the Correct Direction", Science, vol. 319, pp. 955-958, 2008. http://dx.doi.org/10.1126/science.1151343
- N. Mnatsakanyan, J.A. Hook, L. Quisenberry, and J. Weber, "ATP Synthase with Its γ Subunit Reduced to the N-terminal Helix Can Still Catalyze ATP Synthesis", Journal of Biological Chemistry, vol. 284, pp. 26519-26525, 2009. http://dx.doi.org/10.1074/jbc.M109.030528
- M.D. Hossain, S. Furuike, Y. Maki, K. Adachi, T. Suzuki, A. Kohori, H. Itoh, M. Yoshida, and K. Kinosita, "Neither Helix in the Coiled Coil Region of the Axle of F1-ATPase Plays a Significant Role in Torque Production", Biophysical Journal, vol. 95, pp. 4837-4844, 2008. http://dx.doi.org/10.1529/biophysj.108.140061
- M.D. Hossain, S. Furuike, Y. Maki, K. Adachi, M.Y. Ali, M. Huq, H. Itoh, M. Yoshida, and K. Kinosita, "The Rotor Tip Inside a Bearing of a Thermophilic F1-ATPase Is Dispensable for Torque Generation", Biophysical Journal, vol. 90, pp. 4195-4203, 2006. http://dx.doi.org/10.1529/biophysj.105.079087
- E. Usukura, T. Suzuki, S. Furuike, N. Soga, E. Saita, T. Hisabori, K. Kinosita, and M. Yoshida, "Torque Generation and Utilization in Motor Enzyme F0F1-ATP Synthase", Journal of Biological Chemistry, vol. 287, pp. 1884-1891, 2012. http://dx.doi.org/10.1074/jbc.M111.305938
- J.A. Mayr, V. Havlickova, F. Zimmermann, I. Magler, V. Kaplanova, P. Jesina, A. Pecinova, H. Nuskova, J. Koch, W. Sperl, and J. Houstek, "Mitochondrial ATP synthase deficiency due to a mutation in the ATP5E gene for the F1 subunit", Human Molecular Genetics, vol. 19, pp. 3430-3439, 2010. http://dx.doi.org/10.1093/hmg/ddq254
- D.S. Lieber, S.E. Calvo, K. Shanahan, N.G. Slate, S. Liu, S.G. Hershman, N.B. Gold, B.A. Chapman, D.R. Thorburn, G.T. Berry, J.D. Schmahmann, M.L. Borowsky, D.M. Mueller, K.B. Sims, and V.K. Mootha, "Targeted exome sequencing of suspected mitochondrial disorders", Neurology, vol. 80, pp. 1762-1770, 2013. http://dx.doi.org/10.1212/WNL.0b013e3182918c40
- A.I. Jonckheere, G.H. Renkema, M. Bras, L.P. van den Heuvel, A. Hoischen, C. Gilissen, S.B. Nabuurs, M.A. Huynen, M.C. de Vries, J.A. Smeitink, and R.J. Rodenburg, "A complex V ATP5A1 defect causes fatal neonatal mitochondrial encephalopathy", Brain, vol. 136, pp. 1544-1554, 2013. http://dx.doi.org/10.1093/brain/awt086
- I.J. Holt, A.E. Harding, R.K. Petty, and J.A. Morgan-Hughes, "A new mitochondrial disease associated with mitochondrial DNA heteroplasmy.", American journal of human genetics, 1990. http://www.ncbi.nlm.nih.gov/pubmed/2137962
- S.B. Vik, and R.R. Ishmukhametov, "Structure and Function of Subunit a of the ATP Synthase of Escherichia coli", Journal of Bioenergetics and Biomembranes, vol. 37, pp. 445-449, 2005. http://dx.doi.org/10.1007/s10863-005-9488-6
- J. DeLeon-Rangel, R.R. Ishmukhametov, W. Jiang, R.H. Fillingame, and S.B. Vik, "Interactions between subunits a and b in the rotary ATP synthase as determined by cross‐linking", FEBS Letters, vol. 587, pp. 892-897, 2013. http://dx.doi.org/10.1016/j.febslet.2013.02.012
- J.C. Long, J. DeLeon-Rangel, and S.B. Vik, "Characterization of the First Cytoplasmic Loop of Subunit a of the Escherichia coli ATP Synthase by Surface Labeling, Cross-linking, and Mutagenesis", Journal of Biological Chemistry, vol. 277, pp. 27288-27293, 2002. http://dx.doi.org/10.1074/jbc.M202118200
- K.J. Moore, and R.H. Fillingame, "Structural Interactions between Transmembrane Helices 4 and 5 of Subunit a and the Subunit c Ring of Escherichia coli ATP Synthase", Journal of Biological Chemistry, vol. 283, pp. 31726-31735, 2008. http://dx.doi.org/10.1074/jbc.M803848200
- J. Velours, P. Paumard, V. Soubannier, C. Spannagel, J. Vaillier, G. Arselin, and P. Graves, "Organisation of the yeast ATP synthase F0:a study based on cysteine mutants, thiol modification and cross-linking reagents", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1458, pp. 443-456, 2000. http://dx.doi.org/10.1016/S0005-2728(00)00093-1
- C.M. Angevine, K.A. Herold, O.D. Vincent, and R.H. Fillingame, "Aqueous Access Pathways in ATP Synthase Subunit a", Journal of Biological Chemistry, vol. 282, pp. 9001-9007, 2007. http://dx.doi.org/10.1074/jbc.M610848200
- A. Moslemi, N. Darin, M. Tulinius, A. Oldfors, and E. Holme, "Two New Mutations in theMTATP6Gene Associated with Leigh Syndrome", Neuropediatrics, vol. 36, pp. 314-318, 2005. http://dx.doi.org/10.1055/s-2005-872845
- A. Childs, T. Hutchin, K. Pysden, L. Highet, J. Bamford, J. Livingston, and Y. Crow, "Variable Phenotype Including Leigh Syndrome with a 9185T>C Mutation in the MTATP6 Gene", Neuropediatrics, vol. 38, pp. 313-316, 2007. http://dx.doi.org/10.1055/s-2008-1065355
- A.E. Castagna, J. Addis, R.R. McInnes, J.T. Clarke, P. Ashby, S. Blaser, and B.H. Robinson, "Late onset Leigh syndrome and ataxia due to a T to C mutation at bp 9,185 of mitochondrial DNA", American Journal of Medical Genetics Part A, vol. 143A, pp. 808-816, 2007. http://dx.doi.org/10.1002/ajmg.a.31637
- C.M. VERITY, A.M. WINSTONE, L. STELLITANO, D. KRISHNAKUMAR, R. WILL, and R. McFARLAND, "The clinical presentation of mitochondrial diseases in children with progressive intellectual and neurological deterioration: a national, prospective, population‐based study", Developmental Medicine & Child Neurology, vol. 52, pp. 434-440, 2010. http://dx.doi.org/10.1111/j.1469-8749.2009.03463.x
- N. Gigarel, L. Hesters, D. Samuels, S. Monnot, P. Burlet, V. Kerbrat, F. Lamazou, A. Benachi, R. Frydman, J. Feingold, A. Rotig, A. Munnich, J. Bonnefont, N. Frydman, and J. Steffann, "Poor Correlations in the Levels of Pathogenic Mitochondrial DNA Mutations in Polar Bodies versus Oocytes and Blastomeres in Humans", The American Journal of Human Genetics, vol. 88, pp. 494-498, 2011. http://dx.doi.org/10.1016/j.ajhg.2011.03.010
- L. Pereira, P. Soares, P. Radivojac, B. Li, and D. Samuels, "Comparing Phylogeny and the Predicted Pathogenicity of Protein Variations Reveals Equal Purifying Selection across the Global Human mtDNA Diversity", The American Journal of Human Genetics, vol. 88, pp. 433-439, 2011. http://dx.doi.org/10.1016/j.ajhg.2011.03.006
- G. Pfeffer, E.L. Blakely, C.L. Alston, A. Hassani, M. Boggild, R. Horvath, D.C. Samuels, R.W. Taylor, and P.F. Chinnery, "Adult-onset spinocerebellar ataxia syndromes due toMTATP6mutations", Journal of Neurology, Neurosurgery & Psychiatry, vol. 83, pp. 883-886, 2012. http://dx.doi.org/10.1136/jnnp-2012-302568
- K. Auré, O. Dubourg, C. Jardel, L. Clarysse, D. Sternberg, E. Fournier, P. Laforêt, N. Streichenberger, P. Petiot, H. Gervais-Bernard, C. Vial, A. Bedat-Millet, V. Drouin-Garraud, F. Bouillaud, C. Vandier, B. Fontaine, and A. Lombès, "Episodic weakness due to mitochondrial DNA MT-ATP6/8 mutations", Neurology, vol. 81, pp. 1810-1818, 2013. http://dx.doi.org/10.1212/01.wnl.0000436067.43384.0b
- R.P. Saneto, and K.K. Singh, "Illness-induced exacerbation of Leigh syndrome in a patient with the MTATP6 mutation, m. 9185 T>C", Mitochondrion, vol. 10, pp. 567-572, 2010. http://dx.doi.org/10.1016/j.mito.2010.05.006
- R. Carrozzo, J. Murray, O. Capuano, A. Tessa, G. Chichierchia, M. Neglia, R. Capaldi, and F. Santorelli, "A novel mtDNA mutation in the ATPase6 gene studied by E. coli modeling", Neurological Sciences, vol. 21, pp. S983-S984, 2000. http://dx.doi.org/10.1007/s100720070016
- R. Carrozzo, J. Murray, F. Santorelli, and R. Capaldi, "The T9176G mutation of human mtDNA gives a fully assembled but inactive ATP synthase when modeled in Escherichia coli", FEBS Letters, vol. 486, pp. 297-299, 2000. http://dx.doi.org/10.1016/S0014-5793(00)02244-4
- V. Álvarez-Iglesias, F. Barros, �. Carracedo, and A. Salas, "Minisequencing mitochondrial DNA pathogenic mutations", BMC Medical Genetics, vol. 9, 2008. http://dx.doi.org/10.1186/1471-2350-9-26
- D. Thyagarajan, S. Shanske, M. Vazquez ‐Memije, D. Devivo, and S. Dimauro, "A novel mitochondrial ATPase 6 point mutation in familial bilateral striatal necrosis", Annals of Neurology, vol. 38, pp. 468-472, 1995. http://dx.doi.org/10.1002/ana.410380321
- Y. Campos, M.A. Martín, J.C. Rubio, L.G. Solana, C. García-Benayas, J.L. Terradas, and J. Arenas, "Leigh syndrome associated with the T9176C mutation in the ATPase 6 gene of mitochondrial DNA", Neurology, vol. 49, pp. 595-597, 1997. http://dx.doi.org/10.1212/WNL.49.2.595
- C. Dionisi‐Vici, S. Seneca, M. Zeviani, G. Fariello, M. Rimoldi, E. Bertini, and L. De Meirleir, "Fulminant Leigh syndrome and sudden unexpected death in a family with the T9176C mutation of the mitochondrial ATPase 6 gene", Journal of Inherited Metabolic Disease, vol. 21, pp. 2-8, 1998. http://dx.doi.org/10.1023/A:1005397227996
- M. Makino, S. Horai, Y. Goto, and I. Nonaka, "Confirmation that a T-to-C mutation at 9176 in mitochondrial DNA is an additional candidate mutation for Leigh's syndrome", Neuromuscular Disorders, vol. 8, pp. 149-151, 1998. http://dx.doi.org/10.1016/S0960-8966(98)00017-0
- M.E. Rubio-Gozalbo, K.P. Dijkman, L.P. van den Heuvel, R.C. Sengers, U. Wendel, and J.A. Smeitink, "Clinical differences in patients with mitochondriocytopathies due to nuclear versus mitochondrial DNA mutations.", Human mutation, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10862082
- D. Mishmar, and I. Zhidkov, "Evolution and disease converge in the mitochondrion", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1797, pp. 1099-1104, 2010. http://dx.doi.org/10.1016/j.bbabio.2010.01.003
- K. Nakano, "Continuous culture of novel mitochondrial cells lacking nuclei", Mitochondrion, vol. 3, pp. 21-27, 2003. http://dx.doi.org/10.1016/S1567-7249(03)00056-4
- S.F. Dobrowolski, J. Gray, T. Miller, and M. Sears, "Identifying sequence variants in the human mitochondrial genome using high-resolution melt (HRM) profiling", Human Mutation, vol. 30, pp. 891-898, 2009. http://dx.doi.org/10.1002/humu.21003
- Y. Nishigaki, H. Ueno, J. Coku, Y. Koga, T. Fujii, K. Sahashi, K. Nakano, M. Yoneda, M. Nonaka, L. Tang, C. Liou, V. Paquis-Flucklinger, Y. Harigaya, T. Ibi, Y. Goto, H. Hosoya, S. DiMauro, M. Hirano, and M. Tanaka, "Extensive screening system using suspension array technology to detect mitochondrial DNA point mutations", Mitochondrion, vol. 10, pp. 300-308, 2010. http://dx.doi.org/10.1016/j.mito.2010.01.003
- R. Kucharczyk, N. Ezkurdia, E. Couplan, V. Procaccio, S.H. Ackerman, M. Blondel, and J. di Rago, "Consequences of the pathogenic T9176C mutation of human mitochondrial DNA on yeast mitochondrial ATP synthase", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1797, pp. 1105-1112, 2010. http://dx.doi.org/10.1016/j.bbabio.2009.12.022
- Y. Shidara, K. Yamagata, T. Kanamori, K. Nakano, J.Q. Kwong, G. Manfredi, H. Oda, and S. Ohta, "Positive Contribution of Pathogenic Mutations in the Mitochondrial Genome to the Promotion of Cancer by Prevention from Apoptosis", Cancer Research, vol. 65, pp. 1655-1663, 2005. http://dx.doi.org/10.1158/0008-5472.CAN-04-2012
- C. Verny, N. Guegen, V. Desquiret, A. Chevrollier, A. Prundean, F. Dubas, J. Cassereau, M. Ferre, P. Amati-Bonneau, D. Bonneau, P. Reynier, and V. Procaccio, "Hereditary spastic paraplegia-like disorder due to a mitochondrial ATP6 gene point mutation", Mitochondrion, vol. 11, pp. 70-75, 2011. http://dx.doi.org/10.1016/j.mito.2010.07.006
- D. Ronchi, A. Bordoni, A. Cosi, M. Rizzuti, E. Fassone, A. Di Fonzo, M. Servida, M. Sciacco, M. Collotta, M. Ronzoni, V. Lucchini, M. Mattioli, M. Moggio, N. Bresolin, S. Corti, and G.P. Comi, "Unusual adult-onset Leigh syndrome presentation due to the mitochondrial m.9176T>C mutation", Biochemical and Biophysical Research Communications, vol. 412, pp. 245-248, 2011. http://dx.doi.org/10.1016/j.bbrc.2011.07.076
- E. Ciafaloni, F.M. Santorelli, S. Shanske, T. Deonna, E. Roulel, C. Janzer, G. Pescia, and S. DiMauro, "Maternally inherited Leigh syndrome", The Journal of Pediatrics, vol. 122, pp. 419-422, 1993. http://dx.doi.org/10.1016/S0022-3476(05)83431-6
- M.E. Vázquez‐Memije, S. Shanske, F.M. Santorelli, P. Kranz‐Eble, D.C. DeVivo, and S. DiMauro, "Comparative biochemical studies of ATPases in cells from patients with the T8993G or T8993C mitochondrial DNA mutations", Journal of Inherited Metabolic Disease, vol. 21, pp. 829-836, 1998. http://dx.doi.org/10.1023/A:1005418718299
- D.D. de Vries, B.G.M. van Engelen, F.J.M. Gabreëls, W. Ruitenbeek, and B.A. van Oost, "A second missense mutation in the mitochondrial ATPase 6 gene in Leigh's syndrome", Annals of Neurology, vol. 34, pp. 410-412, 1993. http://dx.doi.org/10.1002/ana.410340319
- F.M. Santorelli, S. Shanske, K.D. Jain, D. Tick, E.A. Schon, and S. DiMauro, "A T → C mutation at nt 8993 of mitochondrial DNA in a child with Leigh syndrome", Neurology, vol. 44, pp. 972-972, 1994. http://dx.doi.org/10.1212/WNL.44.5.972
- F. PALLOTTI, A. BARACCA, E. HERNANDEZ-ROSA, W. WALKER, G. SOLAINI, G. LENAZ, G. MELZI d'ERIL, S. DiMAURO, E. SCHON, and M. DAVIDSON, "Biochemical analysis of respiratory function in cybrid cell lines harbouring mitochondrial DNA mutations", Biochemical Journal, vol. 384, pp. 287-293, 2004. http://dx.doi.org/10.1042/BJ20040561
- E. Morava, R.J. Rodenburg, F. Hol, M. de Vries, A. Janssen, L. van den Heuvel, L. Nijtmans, and J. Smeitink, "Clinical and biochemical characteristics in patients with a high mutant load of the mitochondrial T8993G/C mutations", American Journal of Medical Genetics Part A, vol. 140A, pp. 863-868, 2006. http://dx.doi.org/10.1002/ajmg.a.31194
- M.T. Rantamäki, H.K. Soini, S.M. Finnilä, K. Majamaa, and B. Udd, "Adult‐onset ataxia and polyneuropathy caused by mitochondrial 8993T→C mutation", Annals of Neurology, vol. 58, pp. 337-340, 2005. http://dx.doi.org/10.1002/ana.20555
- A.E. Harding, I.J. Holt, M.G. Sweeney, M. Brockington, and M.B. Davis, "Prenatal diagnosis of mitochondrial DNA8993 T----G disease.", American journal of human genetics, 1992. http://www.ncbi.nlm.nih.gov/pubmed/1539598
- G.M. Pastores, F.M. Santorelli, S. Shanske, B.D. Gelb, B. Fyfe, D. Wolfe, and J.P. Willner, "Leigh syndrome and hypertrophic cardiomyopathy in an infant with a mitochondrial DNA point mutation (T8993G)", American Journal of Medical Genetics, vol. 50, pp. 265-271, 1994. http://dx.doi.org/10.1002/ajmg.1320500310
- F.M. Santorelli, S. Shanske, A. Macaya, D.C. DeVivo, and S. DiMauro, "The mutation at nt 8993 of mitochondrial DNA is a common cause of Leigh's syndrome", Annals of Neurology, vol. 34, pp. 827-834, 1993. http://dx.doi.org/10.1002/ana.410340612
- J.M. Shoffner, P.M. Fernhoff, N.S. Krawiecki, D.B. Caplan, P.J. Holt, D.A. Koontz, Y. Takei, N.J. Newman, R.G. Ortiz, M. Polak, S.W. Ballinger, M.T. Lott, and D.C. Wallace, "Subacute necrotizing encephalopathy", Neurology, vol. 42, pp. 2168-2168, 1992. http://dx.doi.org/10.1212/WNL.42.11.2168
- G. Sgarbi, A. Baracca, G. Lenaz, L. Valentino, V. Carelli, and G. Solaini, "Inefficient coupling between proton transport and ATP synthesis may be the pathogenic mechanism for NARP and Leigh syndrome resulting from the T8993G mutation in mtDNA", Biochemical Journal, vol. 395, pp. 493-500, 2006. http://dx.doi.org/10.1042/BJ20051748
- A. Baracca, G. Sgarbi, M. Mattiazzi, G. Casalena, E. Pagnotta, M.L. Valentino, M. Moggio, G. Lenaz, V. Carelli, and G. Solaini, "Biochemical phenotypes associated with the mitochondrial ATP6 gene mutations at nt8993", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1767, pp. 913-919, 2007. http://dx.doi.org/10.1016/j.bbabio.2007.05.005
- S. Mak, C. Chi, C. Liu, C. Pang, and Y. Wei, "Leigh syndrome associated with mitochondrial DNA 8993 T → G mutation and ragged-red fibers", Pediatric Neurology, vol. 15, pp. 72-75, 1996. http://dx.doi.org/10.1016/0887-8994(96)00126-9
- P.R. Steed, and R.H. Fillingame, "Aqueous Accessibility to the Transmembrane Regions of Subunit c of the Escherichia coli F1F0 ATP Synthase", Journal of Biological Chemistry, vol. 284, pp. 23243-23250, 2009. http://dx.doi.org/10.1074/jbc.m109.002501
- E.A. Schon, S. Santra, F. Pallotti, and M.E. Girvin, "Pathogenesis of primary defects in mitochondrial ATP synthesis", Seminars in Cell & Developmental Biology, vol. 12, pp. 441-448, 2001. http://dx.doi.org/10.1006/scdb.2001.0281
- R. Kucharczyk, M. Zick, M. Bietenhader, M. Rak, E. Couplan, M. Blondel, S. Caubet, and J. di Rago, "Mitochondrial ATP synthase disorders: Molecular mechanisms and the quest for curative therapeutic approaches", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1793, pp. 186-199, 2009. http://dx.doi.org/10.1016/j.bbamcr.2008.06.012
- J. Houštěk, T. Mráček, A. Vojtı́šková, and J. Zeman, "Mitochondrial diseases and ATPase defects of nuclear origin", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1658, pp. 115-121, 2004. http://dx.doi.org/10.1016/j.bbabio.2004.04.012
- M. Rak, E. Tetaud, F. Godard, I. Sagot, B. Salin, S. Duvezin-Caubet, P.P. Slonimski, J. Rytka, and J. di Rago, "Yeast Cells Lacking the Mitochondrial Gene Encoding the ATP Synthase Subunit 6 Exhibit a Selective Loss of Complex IV and Unusual Mitochondrial Morphology", Journal of Biological Chemistry, vol. 282, pp. 10853-10864, 2007. http://dx.doi.org/10.1074/jbc.M608692200
- I.C. Soto, F. Fontanesi, M. Valledor, D. Horn, R. Singh, and A. Barrientos, "Synthesis of cytochrome c oxidase subunit 1 is translationally downregulated in the absence of functional F1F0-ATP synthase", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1793, pp. 1776-1786, 2009. http://dx.doi.org/10.1016/j.bbamcr.2009.09.002
- P.E. Hartzog, and B.D. Cain, "The aleu207-->arg mutation in F1F0-ATP synthase from Escherichia coli. A model for human mitochondrial disease.", The Journal of biological chemistry, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8509361
- P. Cortés-Hernández, M.E. Vázquez-Memije, and J.J. García, "ATP6 Homoplasmic Mutations Inhibit and Destabilize the Human F1F0-ATP Synthase without Preventing Enzyme Assembly and Oligomerization", Journal of Biological Chemistry, vol. 282, pp. 1051-1058, 2007. http://dx.doi.org/10.1074/jbc.M606828200
- R. Kucharczyk, M. Rak, and J. di Rago, "Biochemical consequences in yeast of the human mitochondrial DNA 8993T>C mutation in the ATPase6 gene found in NARP/MILS patients", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1793, pp. 817-824, 2009. http://dx.doi.org/10.1016/j.bbamcr.2009.02.011
- M. Rak, E. Tetaud, S. Duvezin-Caubet, N. Ezkurdia, M. Bietenhader, J. Rytka, and J. di Rago, "A Yeast Model of the Neurogenic Ataxia Retinitis Pigmentosa (NARP) T8993G Mutation in the Mitochondrial ATP Synthase-6 Gene", Journal of Biological Chemistry, vol. 282, pp. 34039-34047, 2007. http://dx.doi.org/10.1074/jbc.M703053200
- A.I. Jonckheere, M. Hogeveen, L.G.J. Nijtmans, M.A.M. van den Brand, A.J.M. Janssen, J.H.S. Diepstra, F.C.A. van den Brandt, L.P. van den Heuvel, F.A. Hol, T.G.J. Hofste, L. Kapusta, U. Dillmann, M.G. Shamdeen, J.A.M. Smeitink, and R.J.T. Rodenburg, "A novel mitochondrial ATP8 gene mutation in a patient with apical hypertrophic cardiomyopathy and neuropathy", Journal of Medical Genetics, vol. 45, pp. 129-133, 2007. http://dx.doi.org/10.1136/jmg.2007.052084
- S.M. Ware, N. El-Hassan, S.G. Kahler, Q. Zhang, . Y-W, E. Miller, B. Wong, R.L. Spicer, W.J. Craigen, B.A. Kozel, D.K. Grange, and L. Wong, "Infantile cardiomyopathy caused by a mutation in the overlapping region of mitochondrial ATPase 6 and 8 genes", Journal of Medical Genetics, vol. 46, pp. 308-314, 2009. http://dx.doi.org/10.1136/jmg.2008.063149
- C.J. Ketchum, M.K. Al-Shawi, and R.K. Nakamoto, "Intergenic suppression of the gammaM23K uncoupling mutation in F0F1 ATP synthase by betaGlu-381 substitutions: the role of the beta380DELSEED386 segment in energy coupling.", The Biochemical journal, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9480879
- M.K. Al-Shawi, and R.K. Nakamoto, "Mechanism of Energy Coupling in the FOF1-ATP Synthase: The Uncoupling Mutation, γM23K, Disrupts the Use of Binding Energy To Drive Catalysis", Biochemistry, vol. 36, pp. 12954-12960, 1997. http://dx.doi.org/10.1021/bi971477z
- M.K. Al-Shawi, C.J. Ketchum, and R.K. Nakamoto, "The Escherichia coli FOF1 γM23K Uncoupling Mutant Has a Higher K0.5 for Pi. Transition State Analysis of This Mutant and Others Reveals That Synthesis and Hydrolysis Utilize the Same Kinetic Pathway", Biochemistry, vol. 36, pp. 12961-12969, 1997. http://dx.doi.org/10.1021/bi971478r
ACKNOWLEDGMENTS
The work was supported by a grant from NIH: R01GM66223. Thanks to Drs. John Walker and Andrew Leslie for their past support and discussions, Drs. Alexander Tzagoloff and Rosemary Stuart for their discussions and thoughtful insights, Dr. Vamsi Mootha for his collaborative interactions and editing the manuscript, Dr. Jindrich Symersky his past and current collaborative interactions.
COPYRIGHT
© 2015

Understanding structure, function, and mutations in the mitochondrial ATP synthase by Ting Xu et al. is licensed under a Creative Commons Attribution 4.0 International License.








