
Research Reports:
Microbial Cell, Vol. 9, No. 1, pp. 1 - 20; doi: 10.15698/mic2022.01.767
Detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its first variants in fourplex real-time quantitative reverse transcription-PCR assays
1 Plateforme RNomique et de Génomique Fonctionnelle, Université de Sherbrooke, Sherbrooke, QC, Canada.
2 Département de Microbiologie et d’Infectiologie, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, QC, Canada.
3 Laboratoire de Microbiologie, Centre Intégré Universitaire de Santé et de Services Sociaux (CIUSSS) de l’Estrie, Centre Hospitalier Universitaire de Sherbrooke (CHUS), Sherbrooke, QC, Canada.
4 Département de Biochimie et de Génomique Fonctionnelle, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, QC, Canada.
Keywords: COVID-19, SARS-CoV-2, genetic variants, real-time TaqMan reverse transcription PCR assays, molecular diagnostics, locked nucleic acid (LNA).
Received originally: 20/09/2021 Received in revised form: 17/11/2021
Accepted: 22/11/2021
Published: 25/11/2021
Correspondence:
Simon Labbé, Département de Biochimie et de Génomique Fonctionnelle, Faculté de médecine et des sciences de la santé, Université de Sherbrooke, Sherbrooke, QC, Canada; Simon.Labbe@USherbrooke.ca
Conflict of interest statement: The authors declare that they have no conflict of inter-est with the content of this article.
Please cite this article as: Mathieu Durand, Philippe Thibault, Simon Lévesque, Ariane Brault, Alex Carignan, Louis Valiquette, Philippe Martin and Si-mon Labbé (2021). Detection of severe acute respirato-ry syndrome coronavirus 2 (SARS-CoV-2) and its first variants in fourplex real-time quantitative reverse transcription-PCR assays. Microbial Cell 9(1): 1-20. doi: 10.15698/mic2022.01.767
Abstract
The early diagnosis of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infections is required to identify and isolate contagious patients to prevent further transmission of SARS-CoV-2. In this study, we present a multitarget real-time TaqMan reverse transcription PCR (rRT-PCR) assay for the quantitative detection of SARS-CoV-2 and some of its circulating variants harboring mutations that give the virus a selective advantage. Seven different primer-probe sets that included probes containing locked nucleic acid (LNA) nucleotides were designed to amplify specific wild-type and mutant sequences in Orf1ab, Envelope (E), Spike (S), and Nucleocapsid (N) genes. Furthermore, a newly developed primer-probe set targeted human β2-microglobulin (B2M) as a highly sensitive internal control for RT efficacy. All singleplex and fourplex assays detected £ 14 copies/reaction of quantified synthetic RNA transcripts, with a linear amplification range of nine logarithmic orders. Primer-probe sets for detection of SARS-CoV-2 exhibited no false-positive amplifications with other common respiratory pathogens, including human coronaviruses NL63, 229E, OC43, and HKU-1. Fourplex assays were evaluated using 160 clinical samples positive for SARS-CoV-2. Results showed that SARS-CoV-2 viral RNA was detected in all samples, including viral strains harboring mutations in the Spike coding sequence that became dominant in the pandemic. Given the emergence of SARS-CoV-2 variants and their rapid spread in some populations, fourplex rRT-PCR assay containing four primer-probe sets represents a reliable approach to allow quicker detection of circulating relevant variants in a single reaction.
INTRODUCTION
The human severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that causes the coronavirus disease 2019 (COVID-19) started in the Chinese city of Wuhan in late 2019, and then spread around the world [1][2][3]. This novel coronavirus belongs to the subgroup betacoronavirus and the subgenus Sarbecovirus [3][4][5]. The SARS-CoV-2 genome is constituted of a positive single-stranded RNA with a genome size of nearly ~29,800 to 29,900 nucleotides in length [5]. At the beginning of the outbreak, numerous patients exhibiting an atypical viral pneumonia in Wuhan City were notified to World Health Organization [1]. Shortly thereafter, infected foreign residents of China and foreign travelers arriving in their countries of residence from international destinations, including visitors from Wuhan, contributed to the spread of the virus through person-to-person contact [4][6][7]. Subsequently, spreading of the virus occurred easily and rapidly from person-to-person transmission in local communities, resulting in a pandemic spread across the globe.
–
Although a number of vaccines against SARS-CoV-2 have been developed, they are not available to all people as their production and distribution require efficient logistic and time. Until vaccines become universally available, governments from many countries around the world strongly suggest to their citizens to maintain and sometimes enforce social distancing as they impose lockdown measures, overnight curfew and implement an obligatory quarantine in the case of international travelers flying or driving to different countries. Although rapid identification of SARS-CoV-2-infected patients is critical to trigger off their isolation, the early diagnosis of SARS-CoV-2 remains difficult for the following reasons. First, some SARS-CoV-2-infected patients exhibit no symptoms throughout the course of the infection. Second, other SARS-CoV-2-infected patients with mild symptoms could be confounded with patients who possess other kinds of atypical respiratory tract infections [8][9][10][11]. In response to the outbreak of COVID-19, numerous national public health agencies across the world have been proactive by setting up testing programs. Most of these agencies have recommended and used a nucleic-acid-based method for early detection of SARS-CoV-2. These nucleotide-based tests rely on the real-time reverse transcription PCR (rRT-PCR) that has been considered the “gold standard” approach for SARS-CoV-2 detection due to its high sensitivity for correctly identifying the viral RNA genome found in nasopharyngeal swabs and sputum samples collected from infected patients [12][13][14].
–
For daily clinical specimen testing, clinical microbiology laboratories have used different primer-probe sets for detection of SARS-CoV-2 [12][14][15]. Among them, the CDC N2 and Corman E (also called E Sarbeco) primer-probe sets have been identified to be particularly sensitive [15]. A typical singleplex reaction contains one set of primer pairs (forward and reverse) and a TaqMan probe that hybridizes to a specific targeted region of the viral genome. Short sequences within the nucleocapsid (N) and envelope (E) gene regions are examples of viral templates proved to be detectable with high reproducibility [12][14]. In the same master mixture, a second set of primer pairs and TaqMan probe containing a distinct fluorophore are added to target a human gene, such as the ribonuclease P gene (RNase P also denoted RP or RPP30) to monitor nucleic acid extraction and amplification [14][16].
–
Since the beginning of the COVID-19 pandemic, several studies have reported genomic sequence variations of SARS-CoV-2 isolates [17][18][19][20][21][22]. Among these genetic variations, a single nucleotide polymorphism (SNP) found at position 8,782 (in orf1ab; C instead of T) of SARS-CoV-2 genomic sequence is a hallmark of the L strain [23], whereas the unaltered nucleotide is found at the same position in the S strain. Several mutations have also been found in the coding sequence of the Spike protein (S-protein) [24][25]. One critical mutation in which an adenine has been substituted by a guanine at position 23,403 in the genome of the Wuhan reference strain produces a mutant form of Spike containing D614G substitution (aspartic acid to glycine substitution at amino acid residue 614) [26]. Another critical mutation in which an adenine has been substituted by a thymine at position 23,063 generates a mutant form of Spike harboring N501Y substitution (asparagine to tyrosine substitution at amino acid residue 501) that is found in the United Kingdom (UK) B.1.1.7 strain as well as other reported variants [17]. These mutations represent few examples of the ability of SARS-CoV-2 to rapidly evolve since the beginning of the outbreak.
–
In this study, we report the development of novel primer-probe sets for the detection of SARS-CoV-2 variants. We use fluorogenic probes containing locked nucleic acid (LNA) for detection of SNPs in the genome of SARS-CoV-2 using the TaqMan-LNA rRT-PCR method. In addition to the new primer-probe sets, we have developed a fourplex rRT-PCR assay in which four sets of primer pairs and fluorogenic probes are contained in a single reaction master mixture. Furthermore, we have created and used a novel set of primers/probe that detects the ubiquitously expressed human β2-microglobulin (B2M) transcripts (instead of RNase P) as a positive control for monitoring performance of the whole procedure. These include the presence of RNAs in the collected sample, RNA extraction, reverse transcription efficacy, and real-time amplification. Thus, this multitarget rRT-PCR assay will improve our ability to narrow the differential identification of SARS-CoV-2 isolates.
RESULTS
Limits of detection (LoD) with SARS-CoV-2 RNA transcripts in singleplex reactions
gBlocks DNA templates containing a 5′ T7 RNA polymerase promoter sequence were used to produce viral transcripts that corresponded to specific SARS-CoV-2 genomic regions (Fig. S1). These RNA molecules were used as standards for the generation of standard curves to determine the limit of detection (LoD) of each primer-probe set. Oligonucleotide primer pairs and fluorogenic probes are listed in Table 1. In the case of the E Sarbeco probe and primers, their nucleotide sequences were identical to those described previously [12]. The other primer pairs and fluorogenic probes were designed from the SARS-CoV-2 complete genome [1][5]. In the cases of SARS-CoV-2 L and S strains, specific TaqMan LNA probes were designed to discriminate between the SNP found at position 8,782 (in orf1ab; C instead of T) of SARS-CoV-2 genomic sequence as described previously [23]. Similarly, a specific TaqMan LNA probe was synthesized to detect a guanine instead of an adenine at position 23,403 in the Wuhan reference strain [1][5], allowing the identification of the Spike variant harboring a glycine residue instead of an aspartic acid residue at position 614 (for detection and differentiation of the 614G form versus the original D614 form) [27]. An additional mutation found in the UK strain B.1.1.7 was of interest to probe with a specific TaqMan LNA oligonucleotide to discriminate between the SNP found at position 23,063 (in Spike; thymine instead of adenine) where a tyrosine residue instead of an asparagine residue at position 501 was found (for detection and differentiation of the 501Y form versus the original N501 form) [28]. The sensitivity of each set of primer pair and TaqMan probe was first evaluated in singleplex reactions. Tenfold serial dilutions of viral transcript ranging from 1.3 to 1.4 x 109 copies per reaction mixture were tested in triplicate by rRT-PCR assays. Results were analyzed in terms of the Ct value that was defined as the threshold cycle in which a target viral sequence was first detected. Samples with Ct values ≤ 37 were considered to be positive in comparison with background cross-reactivity of the primers and probes in non-template control (NTC) reactions. The positive Ct value of ≤ 37 was comparable with other reported positive Ct values that have been previously reported [29][30][31][32][33]. The highest dilution of transcript that gave a significative Ct value was defined as the LoD for a targeted RNA transcript. Results showed that LoD values ranged from 1.3 to 13 or 14 RNA transcript copies / reaction (Table 2). Linear regression curves were achieved over a 9-log dynamic range, from 1.3 to 1.3 x 109 copies or 1.4 to 1.4 x 109 copies per reaction for all nine probes, with calculated efficient values of 83.3% to 95.1% (Fig. 1).
–
TABLE 1. Oligonucleotide primers and fluorogenic probes used in real-time quantitative RT-PCR assays. |
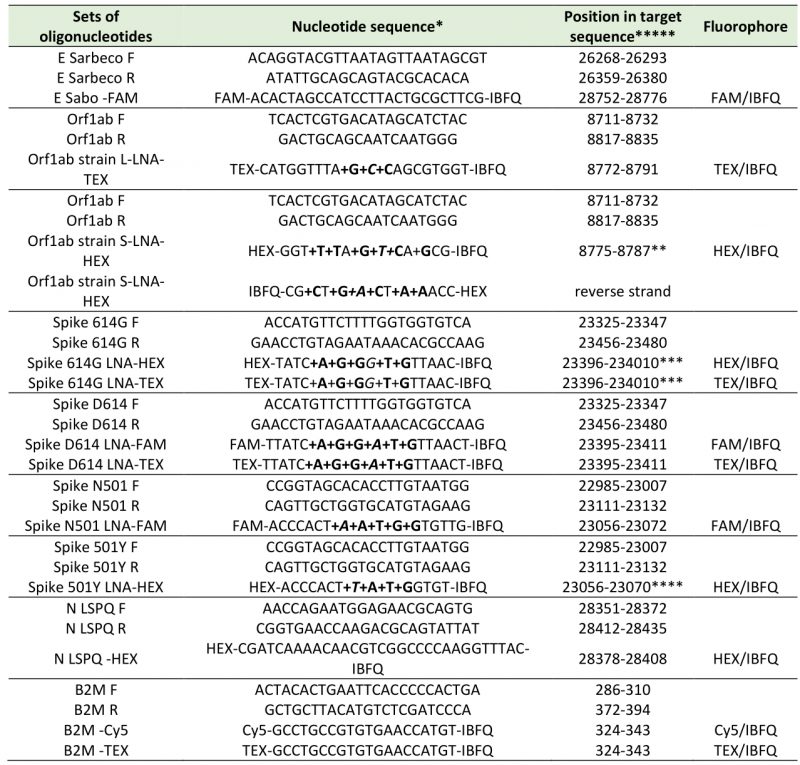 |
*: LNA-modified nucleotides are designated with prefix ‘+’ and are in bold, e.g. +C for a LNA-modified C. **: position with variant mismatch (italic) (C instead of T at position 8782) [23]. ***: position with variant mismatch (italic) (G instead of A at position 23403) [26]. ****: position with variant mismatch (italic) (T instead of A at position 23063) [25]. *****: NC_045512: Reference sequence of SARS-CoV-2 isolate Wuhan-Hu-1, complete genome [1]. NM_004048 : Reference sequence of homo sapiens beta-2-microglobulin (B2M), mRNA. |
–
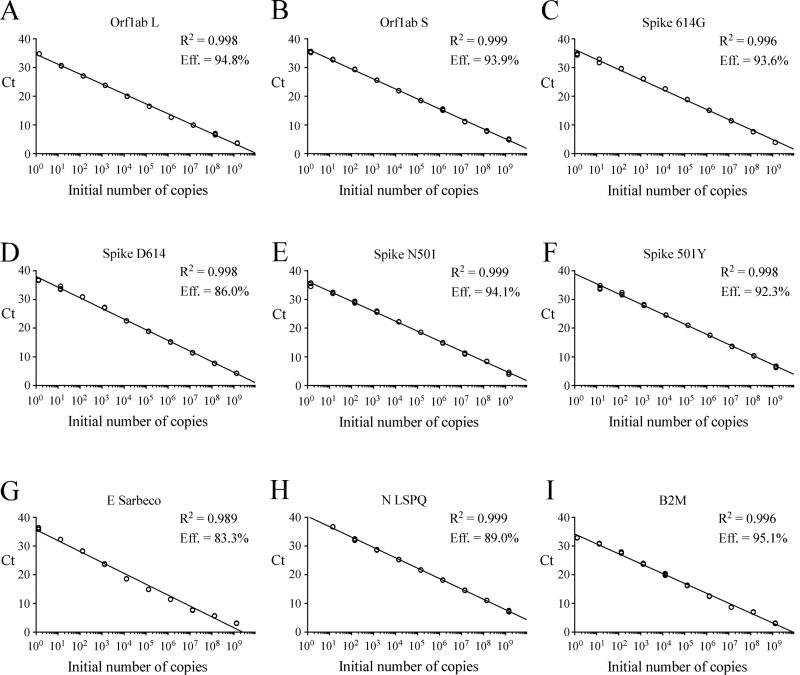 | FIGURE 1: Standard curves to determine synthetic RNA copy number. (A–I) Tenfold serial dilutions ranging from 100 to 109 copies of Orf1ab L, Orf1ab S, Spike 614G, Spike D614, Spike N501, Spike 501Y, E Sarbeco (envelope), N LSPQ (nucleocapsid), and B2M synthetic RNA transcripts were analyzed by rRT-PCR assays. Each representative graph was generated by plotting the Ct values (y-axis) and the log of initial number of copies of synthetic transcripts (x-axis). Calculated linear correlation coefficients (R2) and percentage of amplification efficiencies are indicated for each primer-probe set. The graphs represent quantification of the results of three independent experiments. |
–
LoD in the case of fourplex rRT-PCR assays
Following analytical sensitivity of the primers and probes in singleplex reactions, we sought to develop a fourplex rRT-PCR assay that contained four sets of primers and probes within one reaction master mixture. Limits of detection in the indicated fourplex assays were determined to verify whether their sensitivities were comparable to those of singleplex reactions (Tables 2 and 3). Fourplex reactions that were prioritized included TaqMan probes for detection of single nucleotide polymorphisms in the genome of the SARS-CoV-2 strain that had been found in patient samples [17][26]. With the goal of identifying distinct SARS-CoV-2 virus variants, different combinations of primer-probe sets were mixed within one reaction master mixture to specifically detect viral RNA genes that have undergone genetic variations. As an example, a fourplex master mixture contained all four sets of primers and probes to determine whether a SARS-CoV-2 isolate possessed viral RNA that encoded a Spike variant harboring either an Asp614 or a Gly614 (also denoted D614G mutation) [26]. Similarly, additional fourplex master mixtures contained sets of primers and probes to determine whether a SARS-CoV-2 isolate belongs to the L or S strain [23]. Other examples of fourplex reactions included primer-probe sets to identify the SNP found in Spike gene at position 23,063 (thymine instead of adenine) that results in an amino acid substitution N501Y that triggers a stronger interaction of the Spike receptor-binding domain with the human cell-surface receptor angiotensin-converting enzyme 2 (ACE2) [17][18]. This multiplex approach could facilitate tracking of SARS-CoV-2 variants that carry mutations. Serial 10-fold dilutions of SARS-CoV-2 transcripts were produced as described above and were tested using different combinations of sets of primer pairs and fluorogenic probes in one reaction mixture as described in Table 3. Linear amplification was performed over a 9-log dynamic range, from 1.4 to 1.4 x 109 copies per tetraplex reaction for all combined probes (Table 3). Results showed linear regression curves that exhibited calculated efficient values of 85.1% to 110.4% (Fig. 2).
–
TABLE 2. Limits of detection of SARS-CoV-2-specific primers and probes using synthetic RNA transcripts in singleplex rRT-PCR assays. |
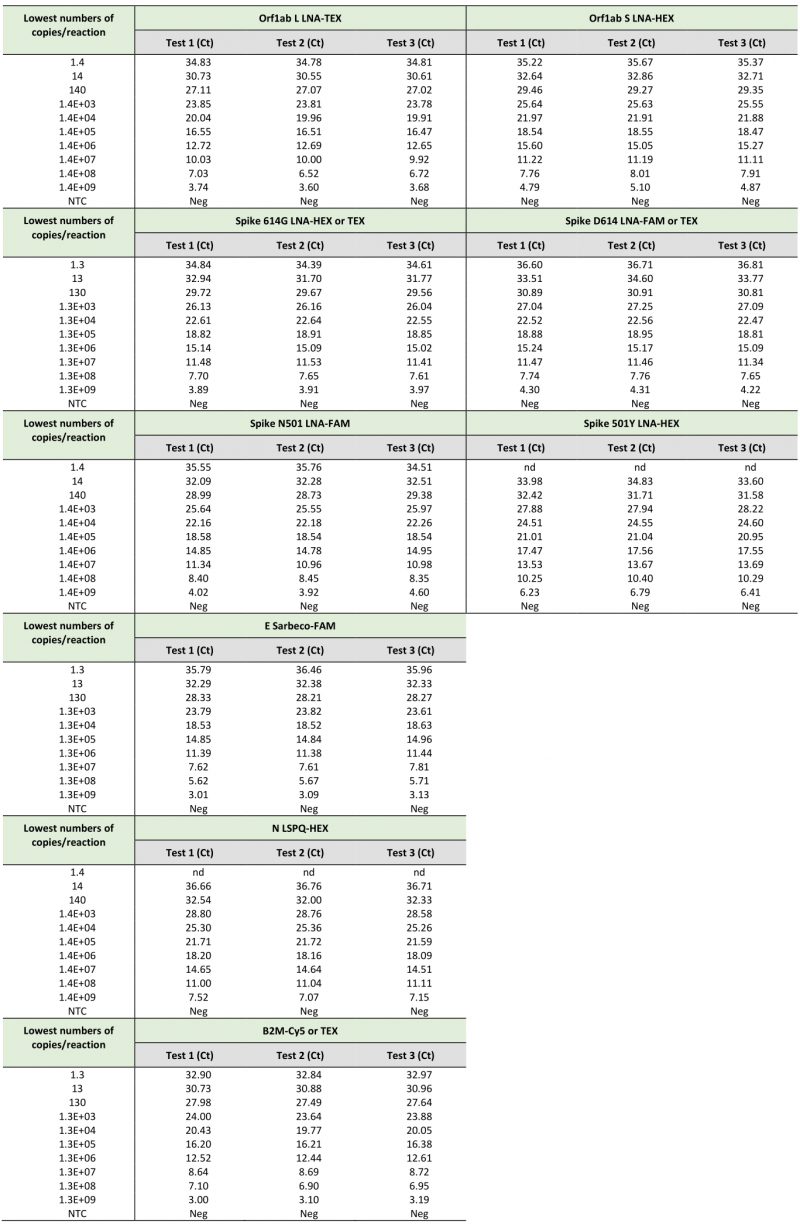 |
–
 | FIGURE 2: Linear regression curves to assess synthetic RNA copy number in fourplex rRT-PCR assays. (A–G) For each representative graph, four primer-probe sets within one reaction mixture were used to detect serial 10-fold dilutions of four synthetic RNA transcripts ranging from 100 to 109 copies per reaction. The y and x axes of each graph are the Ct values and the log of the input of synthetic RNA transcripts. respectively. For each fourplex reaction, R2 indicates calculated linear coefficients and Eff. shows percentage of amplification efficiencies for each primer-probe set that is present with three other sets of primers and probes within one reaction master mixture. Fluorogenic probe color codes are as follows: blue, E Sarbeco; red, Orf1ab L; violet, N LSPQ; black, B2M; green, Spike 614G; orange, Orf1ab S; brown, Spike D614; pink, Spike 501Y; and gold, Spike N501. The graphs represent quantification of the results of three independent experiments. |
–
When we compared the sensitivity measured as the lowest Ct value of the TaqMan probes (Orf1ab L, Orf1ab S, Spike 614G, Spike D614, Spike N501, Spike 501Y, E Sarbeco, N LSPQ, and B2M) in singleplex reactions (Table 2) and fourplex reactions (Table 3), results showed that sensitivities of these probes between singleplex and fourplex reactions were comparable. In the cases of E Sarbeco, Orf1ab L, and B2M, their limit of detection was generally 1.3 or 1.4 copies of transcript per reaction in both singleplex and fourplex rRT-PCR assays. In the cases of Orf1ab S, Spike D614, Spike 614G, Spike N501, Spike 501Y, and N LSPQ probes, a limit of detection of 13 or 14 copies per reaction was observed with much more consistency in singleplex and fourplex assays. Taken together, all designed primer-probe sets performed comparably in singleplex and fourplex assays that reproducibly detected as few as 1.4 to 14 copies of target sequences per reaction.
–
Table 3. Sensitivities of SARS-CoV-2-specific primers and probes in fourplex rRT-PCR assays. |
| Table 3. Sensitivities of SARS-CoV-2-specific primers and probes in fourplex rRT-PCR assays. |
–
Specificity of primers and probes in fourplex reactions
In the course of designing primer and probe sequences used for detection of SARS-CoV-2, we performed BLAST analyses to verify the absence of significant sequence homologies with other respiratory viruses and human genome sequences to avoid the possibility of false positive results. To test the specificity of the primers and probes that we have used in fourplex rRT-PCR reactions, we performed several assays using aliquots of the entire collection of NATtrol Respiratory Verification Panel (NATRVP) from ZeptoMetrix, which contains purified intact respiratory virus and bacteria particles that have been inactivated to make them non-infectious. These microbial particles were supplied in liquid samples containing a specialized matrix that included human cells to mimic the composition of a true clinical specimen. All SARS-CoV-2 primer and probe sets used in fourplex reactions (see Table 3 for all combinations of four sets of primer and probes per one reaction master mixture that have been tested) showed the absence of nonspecific amplification against the ZeptoMetrix NATRVP preparation that includes 19 respiratory pathogens such as influenza A and B, different types of parainfluenza, respiratory syncytial virus A, and other coronaviruses such as NL63, 229E, OC43, and HKU-1 (Table 4). In contrast, the B2M primer-probe set yielded positive results for the same biological samples due to the presence of human cells that are associated with the ZeptoMetrix NATtrol Respiratory pathogen samples. As positive assays for nucleic acid detection of the 19 respiratory pathogens of the ZeptoMetrix NATRVP, we performed BioFire tests using the BioFire FilmArray 2.0 system (BioFire Diagnostics, Salt Lake City, UT). A master mixture containing all viral and bacterial particles of ZeptoMetrix was injected into the BioFire Respiratory Panel 2.1 Pouch that contained all the necessary reagents for automated nuclei acid extraction, reverse transcription, two stages PCR amplification, and detection of multiple respiratory pathogen targets in a single assay. Results showed that the FilmArray instrument in combination with BioFire Pouch reagents including multiple independent sets of primers allowed multiplex-tandem PCR detection of the microbial panel members listed in Table 4. Therefore, FilmArray runs validated the presence of viral and bacterial nucleic acids in the ZeptoMetrix NATRVP preparation. Taken together, the results showed that fourplex assays are specific for the SARS-CoV-2 virus target and that there is an absence of false-positive signals with other respiratory viral or bacterial pathogens.
–
TABLE 4. Specificities of fluorogenic SARS-CoV-2 probes with a number of other viruses and bacteria. |
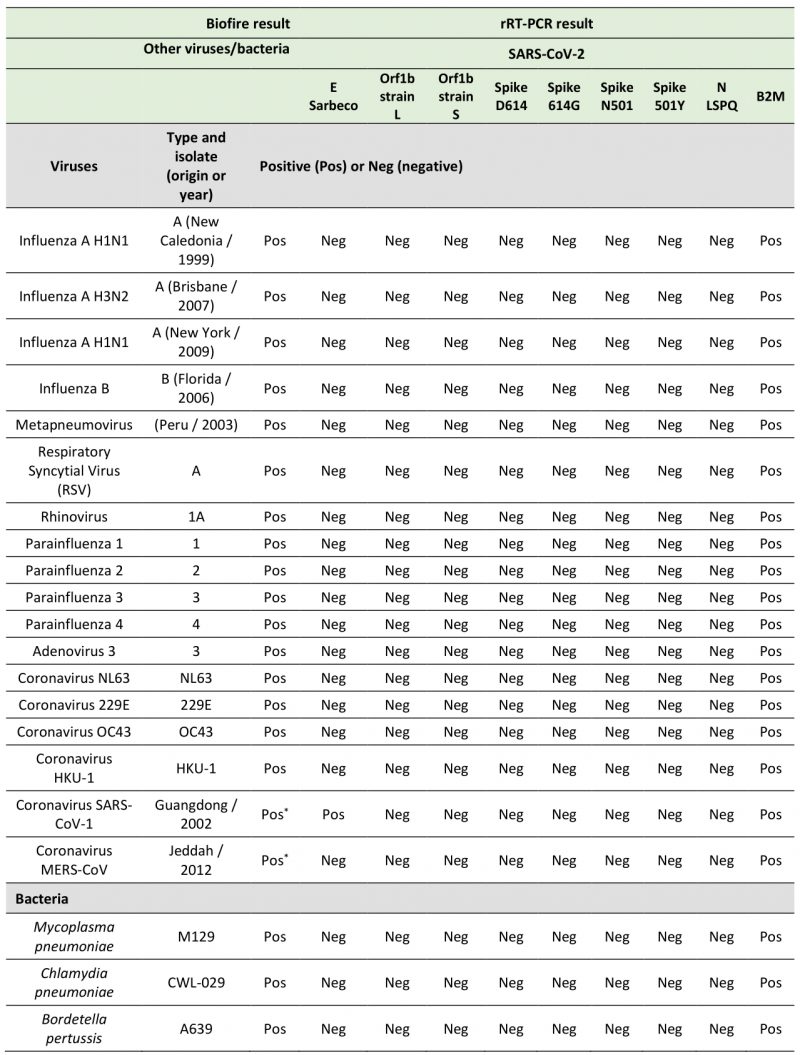 |
* In the two cases marked with an asterisk, results were obtained by bioinformatic analysis and rRT-PCR. |
–
LNA probes allow differential identification of specific SARS-CoV-2 transcripts in fourplex reactions
We tested distinct groups of differentially labeled four-color sets of probes that allow discrimination of single-nucleotide mutations found in genetic variants of SARS-CoV-2 to validate specific LNA probe-target interaction for SNPs detection in the genome of SARS-CoV-2 (Table 5). Results showed that the Orf1ab L probe containing a “C LNA” at the polymorphic site (position 8782 in SARS-CoV-2 sequence; Table 1) generated a positive fluorogenic signal that was specific for transcripts produced from the new circulating L variant strain, whereas no fluorogenic signal was detected in the presence of RNA obtained from the older S variant strain (Table 5; e.g. probes in fourplexes 1 and 2). In the case of the Orf1ab S probe containing a “T LNA” at the polymorphic site (Table 1), this probe exhibited a positive amplification signal in the presence of transcripts produced from the S variant strain and failed to detect transcripts of the L variant strain (Table 5; e.g. probes in fourplexes 3 and 4).
–
TABLE 5. Real-time genotyping with oligonucleotide probes containing locked nucleic acids (LNA) for detection of SARS-CoV-2 variants. |
| TABLE 5. Real-time genotyping with oligonucleotide probes containing locked nucleic acids (LNA) for detection of SARS-CoV-2 variants. |
–
Two oligonucleotide probes containing LNA residues were created for specific hybridization of target sequences containing nucleotide substitutions that confer amino acid substitution in Spike at Asp614 (also denoted D614) and Gly614 (also called 614G; Table 1). The LNA probe for the detection of the Wuhan reference D614 strain carried an “A LNA” at the SNP site (position 23,403 in the viral genome), whereas the fluorogenic probe specific for the detection of the circulating Spike variant (614G) contained a “G LNA” at this position (Table 1). In the case of the Spike D614 LNA probe, results showed that it was specific for the detection of transcripts from the Spike D614 gene and did not generate amplification signal in the presence of transcripts of the Spike 614G variant gene (Table 5; e.g. probes in fourplexes 3 and 5). In contrast, results showed that the Spike 614G LNA probe exhibited high specificity for detection of transcripts from the Spike 614G gene but failed to amplify transcripts from the Spike D614 coding sequence (Table 5; e.g. probes in fourplex 5). Similarly, experiments by fourplex rRT-PCR assays verified that the LNA probe for the detection of Spike 501Y transcripts (nucleotide change A23,063T; Tables 1 and 2) failed to detect the Spike N501 transcripts (Table 5, e.g. probes in fourplex 6). Consistently, results showed that the LNA probe for detection of the Spike N501 transcripts (nucleotide A at position 23,063; Tables 1 and 2) did not detect the Spike 501Y transcripts (Table 5, e.g. probes in fourplex 7). In contrast, results showed that the Spike N501 and Spike 501Y LNA probes exhibited high specificity for detection of transcripts from the Spike N501 and Spike 501Y coding sequences, respectively (Table 5, e.g. probes in fourplex 8). Taken together, the results showed that the oligonucleotide probes containing LNA residues can be used for differential identification of SARS-CoV-2 viral templates in which mutations have occurred.
–
Detection of SARS-CoV-2 RNA in clinical specimens
To evaluate the use of different combinations of sets of primer pairs and fluorogenic probes in fourplex reactions for detection of SARS-CoV-2 and its variants in clinical specimens, we analyzed 160 nasopharyngeal swab samples from patients with clinical laboratory-confirmed COVID-19. Results showed that fourplex A containing probes that allow detection of double-nucleotide substitutions in the coding sequence of Spike (causing N-to-Y substitution at position 501 and D-to-G substitution at position 614) identified a total of 49 samples from patients who have been tested between February 21 and March 30, 2021 (Table 6). In this group of samples, the fourplex A allow the detection of all (100%) the SARS-CoV-2 501Y 614G variants when compared to data obtained by sequencing. In the case of fourplex B, sets of primer pairs and probes were combined to detect the Spike D-to-G amino acid change at position 614 whereas the original asparagine at amino acid 501 remained unchanged. A first group of 62 samples carrying the D614G substitution were found between March 19 and March 25, 2020. Furthermore, almost one year later (between February 21 and March 30, 2021), 39 samples harboring the same mutation were detected for a grand total of 101 samples (Table 6). Fourplex C was used to detect the SARS-CoV-2 strain L that possesses a genome encoding N501 and D614 Spike protein. Five specimens among the samples were detected harboring this genotype between March 19 and March 25, 2020 (Table 6). In the case of the Wuhan reference strain S in which the original amino acids at positions 501 and 614 are asparagine and aspartic acid, respectively, its detection was performed using the fourplex D. This strain was detected in five clinical specimens between March 19 and March 25, 2020 (Table 6). All clinical samples were confirmed to be positive with respect to their mutations or reference nucleotides by sequencing after RT-PCR followed by a second-round of PCR amplification. Taken together, the results showed that our fourplex sets of primers/probes can be effectively used in rRT-PCR assays for differential diagnosis of SARS-CoV-2 variants in clinical specimens.
–
TABLE 6. Test results using fourplex sets of primers/probes for the detection of SARS-CoV-2 variants in clinical specimens. |
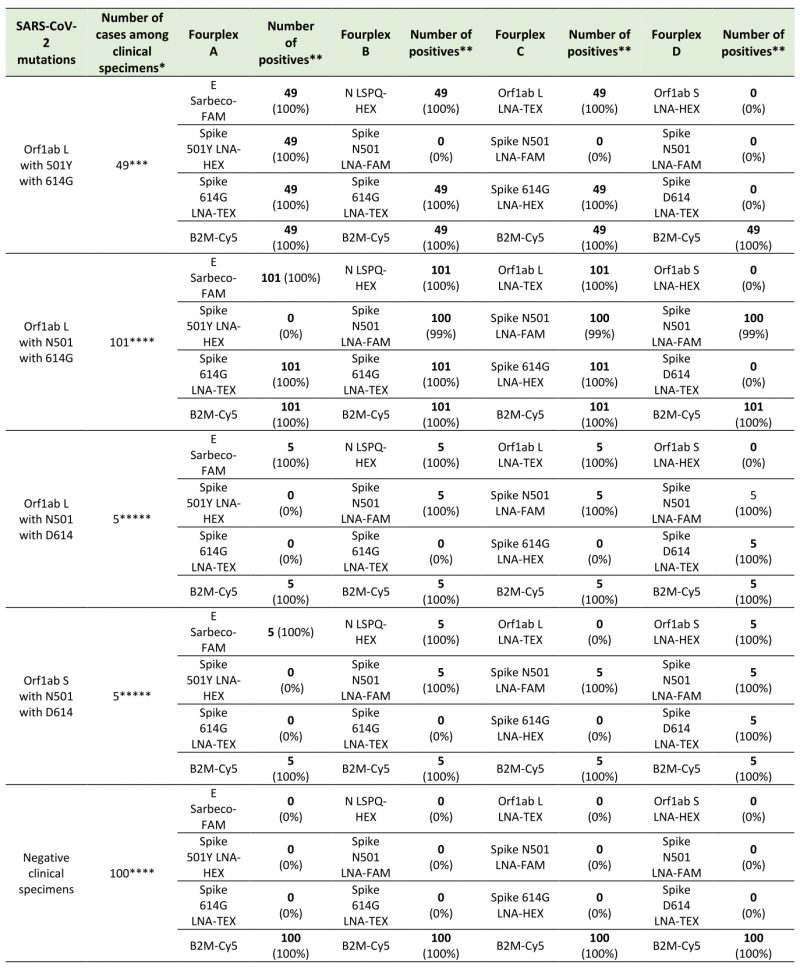 |
* All positives have been confirmed by sequencing ** Number of positive test results (% of detection). *** Sampling period between February 21 and March 30, 2021. **** Sampling periods between March 19 and March 25, 2020 and February 21 and March 30, 2021. ***** Sampling period between March 19 and March 25, 2020. |
DISCUSSION
The ongoing COVID-19 pandemic characterized by successive waves of infection is a driving force for optimization and constant development of SARS-CoV-2 testing assays [13][34]. Here, we report the development of a fourplex TaqMan-LNA rRT-PCR assay that included per single reaction: four sets of primer pairs and four distinct TaqMan probes labelled with different fluorophores. Results showed that a tetraplex reaction in a single master mixture reached a detection limit of 14 viral transcript copies per reaction (Table 3). The amplification efficiencies of tetraplex reactions (values of 85.1% to 110.4%) were equivalent to those in the singleplex reactions (values of 83.3% and 95.1%) in which a single viral primer-probe set was used within one reaction master mixture (Table 2). The primer-probe set targeting a single nucleotide substitution in the Orf1ab L transcript was more sensitive than primer-probe sets targeting single nucleotide substitutions in the Orf1ab S, Spike 614G, Spike D614, Spike N501, and Spike 501Y transcripts. This set could detect transcript at levels as low as 1.4 copies per reaction in singleplex and fourplex rRT-PCR assays (Tables 2 and 3). In the cases of primer-probe sets targeting single nucleotide substitutions in the Orf1ab S, Spike 614G, Spike D614, Spike N501, and Spike 501Y transcripts, a detection limit of 14 transcript copies was reached when they were used in fourplex assays on three independent experiments (Table 3). In the case of the primer-probe set targeting the E gene (encoding the envelope membrane protein E) that is denoted E Sarbeco [12], it was more sensitive than the N LSPQ probe targeting the N gene (encoding the nucleocapsid N protein; Tables 2 and 3). The E Sarbeco primer-probe set could detect transcripts levels as low as 1.3 to 1.4 copies per reaction, whereas 14 transcript copies per reaction was reached with the N LSPQ primer-probe set under our experimental conditions.
–
During the course of studies, we have found that the human B2M gene encoding an ubiquitously expressed protein could be a positive control of choice for monitoring efficiency of RNA extraction from swab samples and subsequent reverse transcription reaction reliability. B2M is classified as one of the top gene most stably expressed with a broad range of expression in the vast majority of cell types and tissues [35]. The B2M transcript exhibits a robust half-life and its abundance is among the highest [36]. Furthermore, B2M is a top reference gene for accurate normalization in RNA expression profiles of human whole blood samples using rRT-qPCR assays [37]. To create the B2M primer-probe set and ensure that its resulting product originates from RNA/complementary DNA (cDNA) amplification and not from genomic DNA amplification, we designed the reverse primer on exon2-exon3 junction of the B2M gene. Because this exon2-exon3 boundary sequence is only present in spliced B2M mRNA and not in genomic DNA, this approach eliminated the possibility that the B2M rRT-PCR signal resulted from an amplification of genomic DNA. Results showed that the B2M primer-probe set was highly sensitive when tested in singleplex assays, exhibiting an efficiency of 95.1% (r2 = 0.996) and a detection limit of 1.3 transcript copies per reaction (Fig. 1 and Table 2). When the B2M primer-probe set was used in fourplex assays, it remained highly sensitive with high levels of rRT-PCR efficiency (95.5% to 106.5%) and a similar detection limit (1.4 transcript copies per reaction) as compared to that observed in the case of singleplex assays (Fig. 1 and 2; Tables 2 and 3). The vast majority of COVID-19 tests are currently targeting the human RNase P gene (denoted RPP30 or h-RP) as a positive control to monitor RNA extraction and its quality to be reverse transcribed for its subsequent PCR amplification [14][16]. However, problems with the use of one CDC-recommended h-RP primer-probe set have been reported previously [38]. One issue is that the reverse primer hybridizes in the same exon as the forward primer and the TaqMan probe, therefore allowing amplification from genomic DNA instead of exclusively from spliced mRNA [38]. In that case, a positive result with this specific primer and probe set fails to validate the quality of the clinical sample specimen with respect to its RNA content.
–
The use of multiplex rRT-PCR assays allowed simultaneous targeting of different regions of the SARS-CoV-2 genome in a single master mixture. Among the targeted regions, we often included the E Sarbeco probe due to its reported successful use and high sensitivity [12][15]. In this manner, Ct values of the E Sarbeco probe in fourplex assays served as a point of reference to assess the significance of Ct values of the other tested probes. Interestingly, we have observed that utilization of multiplex SARS-CoV-2 primer/probe sets with distinct fluorophores shed light on which regions of the virus genome are more sensitive for a rRT-PCR-based SARS-CoV-2-specific identification assay. Another advantage of using a multitarget rRT-PCR method is the fact that it reduces the amount of non-enzymatic reagents, enzyme mixtures and plasticware required to analyze all the samples on a daily basis. We have found that the fourplex assay was three times cheaper as compared to the singleplex assay. This is due to the fact that each fourplex reaction contains three sets of primer pairs and three fluorogenic probes that specifically detect SARS-CoV-2 RNA genes in a single reaction master mixture (the fourth primer-probe set being used to target a human reference transcript such as B2M). In contrast, three times more reagents and three times well plates of thermal cycler(s) are required to obtain the same results using singleplex assays since the duplex approach involves three separate reaction master mixtures.
–
RNA viruses exhibit high mutation rates that may result in self beneficial properties by increasing their ability to infect host cells [39][40][41]. In the case of SARS-CoV-2, several mutations have been identified since the start of the pandemic [21][42][43]. For instance, two linked SNPs in the genome of SARS-CoV-2 have defined two major lineage of the virus that are designated S and L strains [23]. Another example consists of nucleotide mutations found in the viral sequence of the gene encoding Spike that result in increased virus infectivity, especially nucleotide changes resulting in amino acid changes at Asp614 (D614G) and Asn501 (N501Y) [17][26]. Here, we have used forward and reverse primers that hybridize to each side of hot spot mutational sites found in Orf1ab (nucleotide change T8782C, generating lineage S versus L), and at different locations in Spike such as nucleotide change A23403G (resulting in Spike variation D614G) and nucleotide change A23063T (giving rise Spike variation N501Y). These pairs of primers were used in combination with wild-type and mutant-LNA probes that hybridize at the mutation site. The fourplex real-time RT-PCR assay using LNA-based TaqMan probes exploits the 5′-3′ nuclease activity of Taq DNA polymerase, allowing direct detection of each of the four fluorogenic qPCR products by the uncoupling of a reporter dye from its quencher dye during qPCR. Furthermore, LNA modification of TaqMan probes increases their base pairing stability and creates a highly favorable context of duplex formation between them and their target sequences under more stringent conditions [44][45][46][47]. Therefore, these properties allow LNA-based probes to discriminate between distinct viral genomic sequences that differ by a single nucleotide at a precise hot spot mutational site. This was performed, in three different places on the genome of SARS-CoV-2 at the same time into one reaction. In this way, it is possible to detect a range of representative genotypes that could reveal the presence of different variants of SARS-CoV-2 geographically and over time. One limitation of the study is that we need to know in advance the nature of the mutation for a given SARS-CoV-2 variant that we intend to detect. However, once the nature of the mutation is known, we can design the optimal LNA-based probe to discriminate between distinct viral genomic sequences that differ by a single nucleotide at a precise mutational site.
–
Recently, other studies have reported multiplex rRT-PCR assays [48][49]. One study has developed triplex assays to detect SARS-CoV-2 variants of concern but only two new primer-probe sets have been designed, making it difficult to formally identify a specific variant without excluding the presence of additional mutations [48]. Another study has developed a five plex rRT-PCR test that allowed detection of three SARS-CoV-2 target RNA transcripts plus two additional control transcripts [49]. Although five primer-probe sets were designed, several results were obtained using two or three of the viral targets detected with FAM-labelled probes, which was equivalent to perform 3- or 2-plex rRT-PCR assays. One alternative to multiplex rRT-PCR approach consists of performing multi-target loop-mediated isothermal amplification (LAMP) [50]. Although real-time LAMP was specific, this method exhibited lower sensitivity compared to rRT-PCR because none of the LAMP primers were capable of detecting SARS-CoV-2 target genes down to fifty copies on patient samples [50].
–
When we analyzed clinical specimens, we have consistently observed that SARS-CoV-2 strains harboring the Spike gene encoding the original D614 form or belonging to the S lineage have progressively stopped to circulate in the Eastern Townships population of Quebec (Canada) through the month of March 2020 as observed in different regions throughout the world [23][26]. After March 2020, the 614G form as well as the SARS-CoV-2 L lineage became globally dominant. Following the emergence of the SARS-CoV-2 B.1.1.7 variant in the United Kingdom on December 2020 [51][52], within approximately two months, this variant harboring a typical N-to-Y substitution at position 501 of Spike gained prominence in clinical specimens that we have analyzed between February 21 and March 30, 2021. These above-mentioned examples illustrate the effectiveness of our method for differential diagnosis and surveillance of SARS-CoV-2 virus isolates, especially variant of concern or of high consequence as defined by the Center for Disease Control and Prevention (CDC). Furthermore, this multitarget rRT-PCR assay enables rapid follow-up of the most clinically relevant variants across the population in real-time and effectively guide the selection of viral strains that disserve further analysis by their whole-genome sequencing using next-generation sequencing methods.
MATERIALS AND METHODS
Primer and probe design
The Wuhan-Hu-1 genome reference sequence of SARS-CoV-2 [1][5] as well as several other listed sequences of its genome (available in the repository of the National Center for Biotechnology Information) were aligned using BLASTn software to identify conserved nucleotide regions corresponding only to the SARS-CoV-2 viral genome and not found in other viral sequences, including SARS-CoV-1 and MERS-CoV. Subsequently, different primer-probe sets targeting distinct regions of the SARS-CoV-2 genome, especially within E, Orf1ab, and S genes, were designed using Primer Express 3.0.1 software. The N LSPQ primer-probe set was designed as described previously [53]. After validation of their exclusivity by comparing them to Virus Pathogen Resource (ViPR), Reference Viral DataBase (RVDB) and human GRCh38 genomic DNA/mRNA databases, the indicated TaqMan primer-probe sets (Table 1) were predicted, verified, and validated to specifically amplify SARS-CoV-2, exhibiting the absence of non-specific homologies with other respiratory viral pathogens or human-related gene sequences. The only exception was the E Sarbeco primer-probe set that was predicted to hybridize with the E gene sequence of SARS-CoV-1 in addition to the E gene of SARS-CoV-2 [12]. TaqMan probes were labeled at the 5′ end with either 6-carboxyfluorescein (6-FAM), Texas Red 615 (TEX 615), hexachlorofluorescein (HEX), or Cytiva5 (Cy5) and at the 3′ end with either Iowa Black FQ (IBFQ) or Black Hole Quencher 1 (BHQ1) (Integrated DNA Technologies, Coralville, Iowa).
–
In the case of probes containing LNA residues, they were designed for detection of SNPs in the Orf1ab and S genes of SARS-CoV-2. Essentially, we used forward and reverse primers that hybridized with each side of the target sequence and wild-type or mutant TaqMan LNA probes that hybridized at the mutation site, which was located between the forward and reverse primers. All primers and TaqMan probes were designed to avoid self-complementarity and the formation of primer-dimer by-products and hairpins. Furthermore, all primer/probe sets were validated for their compatibility in multiplex rRT-PCR assays. Standard-curve plots for calculation of rRT-PCR amplification efficiencies were performed as described previously [16][54].
–
Synthesis of RNA transcripts used as standards
gBlocks double-stranded DNA fragments encompassing the amplified region of each targeted SARS-CoV-2 region were synthesized accompanied with a T7 promoter sequence at their 5’ends (Fig. S1). A similar approach was used in the case of the DNA fragment covering the amplified region of the human B2M gene. Purified gBlocks products were used for in vitro T7-dependent transcription, as described previously [55]. The gBlocks DNA templates were eliminated by digestion with RNase-free DNase I for 15 min at 37°C. The RNA transcripts were purified using a MEGAclear transcription clean-up kit (Invitrogen) and were quantified spectrophotometrically at 260 nm. After RNA levels quantification, measurements of transcripts were converted to the molecule number per μl as described previously [56]. To validate calculations of viral RNA copies per reaction, we have established standard curves from 10-fold dilutions of RNA isolated from known number of copies from a prototype viral RNA preparation that was commercially available (Exact Diagnostics, Fort Worth, TX; Vircell, Granada, Spain).
–
Real-time quantitative reverse transcription PCR (rRT-PCR)
Reactions were performed in a 10-μl reaction mixture including 7 μl extracted RNA-containing samples or RNA transcripts used as standards, 2.5 μl Reliance One-Step Multiplex rRT-PCR Supermix (4X concentrated) (BioRad), and 0.5 μl of forward and reverse primers (500 nM) and the indicated TaqMan or TaqMan-LNA probe (250 nM). One-step rRT-PCR amplification and detection were performed in a CFX96 Touch Real-Time PCR System (BioRad) under the following conditions. Reverse transcription of 10 min at 50°C was followed by PCR activation at 95°C for 10 min and 50 cycles of amplification (10 s at 95°C and 30 s at 60°C or for some indicated reactions at 63°C). In singleplex reaction mixtures, each reaction mixture contained a single primer pair and TaqMan or TaqMan-LNA probe. In the case of fourplex reactions, they contained all four sets of primers (500 nM per set) and probes (250 nM per probe) within a single reaction master mixture. Fluorescence measurements were monitored after each amplification round and the threshold cycle (Ct) value for each sample was calculated by assessing the point at which fluorescence crossed the threshold line, exhibiting an increase in fluorescence above the calculated background levels. The result was considered valid if two or more of the target-specific fluorescent signals showed the Ct value ≤ 37 cycles and all positive and negative control reactions gave a successful and no amplification, respectively.
–
BioFire FilmArray assays
The collection of respiratory viral and bacterial pathogens was obtained from ZeptoMetrix (cat #NATRVP-IDI). The collection is called NATtrol Respiratory Verification Panel (NATRVP) and contains 20 vials x 0.6 ml, each containing viral and bacterial targets. A master mixture that was constituted of an aliquot of each pathogen of NATRVP was injected into the BioFire Respiratory Panel 2.1 (RP2.1) Pouch in accordance with the manufacturer’s instructions for analyze using the BioFire FilmArray 2.0 System (BioFire Diagnostics) as described previously [57]. The loaded RP2.1 Pouch inserted into the FilmArray instrument contained all necessary reagents for automated nucleic acid extraction, reverse transcription, and two consecutive multiplex PCR amplification runs. Furthermore, the FilmArray instrument has the potential to undergo gene target melt curve analysis with each target in a valid run reported as detected or not detected.
–
Clinical specimens
Deidentified nasopharyngeal swabs from infected individuals were obtained from the Laboratoire de Microbiologie du Centre Intégré Universitaire de Santé et de Services Sociaux de l’Estrie, Centre Hospitalier Universitaire de Sherbrooke. The thermal inactivation of SARS-CoV-2 was performed using 20 μl of nasopharyngeal swab diluent that was mixed with sterile water (20 μl). The sample preparation was heated at 90°C for 2 min and then chilled at 4°C for 3 min. Following this step, 5 μl of heat-inactivated SARS-CoV-2-containing sample was analyzed by direct one-step rRT-PCR as described above in the previous section dedicated to the rRT-PCR method. Positive fourplex rRT-PCR results were confirmed by retesting the samples in singleplex rRT-PCR reactions from which DNA products were separately analyzed by DNA sequencing to confirm identity of each DNA sequence and discriminate between two (or more) viral genomic sequences that differ by a single nucleotide. Aside thermal preparation, we have also performed simultaneous RNA extractions using the MagNA Pure Compact system (Roche). Nucleic acid extracts gave the same results in the case of positive samples of SARS-CoV-2 among the specimens assessed.
–
The authority that provided ethics approval is the Centre Intégré Universitaire de Santé et de Services Sociaux (CIUSSS) de l’Estrie-CHUS. The CIUSSSE-CHUS institutional review board approved this study #2021-4223. The requirement for informed consent was waived because this was a retrospective study that used unmarked and deidentified leftover samples.
REFERENCES
- Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG, Hu Y, Tao ZW, Tian JH, Pei YY, Yuan ML, Zhang YL, Dai FH, Liu Y, Wang QM, Zheng JJ, Xu L, Holmes EC, and Zhang YZ (2020). A new coronavirus associated with human respiratory disease in China. Nature 579:265-269. 10.1038/s41586-020-2008-3
- Zhang YZ and Holmes EC (2020). A genomic perspective on the origin and emergence of SARS-CoV-2. Cell 181:223-227. 10.1016/j.cell.2020.03.035
- Zhu N, Zhang D, Wang W, Li X, Yang B, Song J, Zhao X, Huang B, Shi W, Lu R, Niu P, Zhan F, Ma X, Wang D, Xu W, Wu G, Gao GF, and Tan W (2020). A novel coronavirus from patients with pneumonia in China, 2019. N Engl J Med 382:727-733. 10.1056/NEJMoa2001017
- Chan JF, Kok KH, Zhu Z, Chu H, To KK, Yuan S, and Yuen KY (2020). Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg Microbes Infect 9:221-236. 10.1080/22221751.2020.1719902
- Lu R, Zhao X, Li J, Niu P, Yang B, Wu H, Wang W, Song H, Huang B, Zhu N, Bi Y, Ma X, Zhan F, Wang L, Hu T, Zhou H, Hu Z, Zhou W, Zhao L, Chen J, Meng Y, Wang J, Lin Y, Yuan J, Xie Z, Ma J, Liu W, Wang D, Xu, W, Holmes EC, Gao GF, Wu G, Chen W, Shi W, and Tan W (2020) Genomic characterisation and epidemiology of 2019 novel coronavirus: implications for virus origins and receptor binding. Lancet 395:565-574. 10.1016/s0140-6736(20)30251-8
- Holshue ML, DeBolt C, Lindquist S, Lofy KH, Wiesman J, Bruce H, Spitters C, Ericson K, Wilkerson S, Tural A, Diaz G, Cohn A, Fox L, Patel A, Gerber SI, Kim L, Tong S, Lu X, Lindstrom S, Pallansch MA, Weldon WC, Biggs HM, Uyeki TM, and Pillai SK (2020). First case of 2019 novel coronavirus in the United States. N Engl J Med 382:929-936. 10.1056/NEJMoa2001191
- Oude Munnink BB, Nieuwenhuijse DF, Stein M, O’Toole Á, Haverkate M, Mollers M, Kamga SK, Schapendonk C, Pronk M, Lexmond P, van der Linden A, Bestebroer T, Chestakova I, Overmars RJ, van Nieuwkoop S, Molenkamp R, van der Eijk AA, GeurtsvanKessel C, Vennema H, Meijer A, Rambaut A, van Dissel J, Sikkema RS, Timen A, and Koopmans M (2020). Rapid SARS-CoV-2 whole-genome sequencing and analysis for informed public health decision-making in the Netherlands. Nat Med 26:1405-1410. 10.1038/s41591-020-0997-y
- Guarneri C, Rullo EV, Pavone P, Berretta M, Ceccarelli M, Natale A, and Nunnari G (2021). Silent COVID-19: what your skin can reveal. Lancet Infect Dis 21:24-25. 10.1016/s1473-3099(20)30402-3
- Lai CC, Shih TP, Ko WC, Tang HJ, and Hsueh PR (2020). Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): The epidemic and the challenges. Int J Antimicrob Agents 55:105924. 10.1016/j.ijantimicag.2020.105924
- Rivett L, Sridhar S, Sparkes D, Routledge M, Jones NK, Forrest S, Young J, Pereira-Dias J, Hamilton WL, Ferris M, Torok ME, and Meredith L; CITIID-NIHR COVID-19 BioResource Collaboration, Curran MD, Fuller S, Chaudhry A, Shaw A, Samworth RJ, Bradley JR, Dougan G, Smith KG, Lehner PJ, Matheson NJ, Wright G, Goodfellow IG, Baker S, and Weekes MP (2020). Screening of healthcare workers for SARS-CoV-2 highlights the role of asymptomatic carriage in COVID-19 transmission. Elife 9:e58728. 10.7554/eLife.58728
- Yang R, Gui X, and Xiong Y (2020). Comparison of clinical characteristics of patients with asymptomatic vs symptomatic coronavirus disease 2019 in Wuhan, China. JAMA Netw Open 3:e2010182. 10.1001/jamanetworkopen.2020.10182
- Corman VM, Landt O, Kaiser M, Molenkamp R, Meijer A, Chu DK, Bleicker T, Brünink S, Schneider J, Schmidt ML, Mulders DG, Haagmans BL, van der Veer B, van den Brink S, Wijsman L, Goderski G, Romette JL, Ellis J, Zambon M, Peiris M, Goossens H, Reusken C, Koopmans MP, and Drosten C (2020). Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Euro Surveill 25:2000045. 10.2807/1560-7917.es.2020.25.3.2000045
- Esbin MN, Whitney ON, Chong S, Maurer A, Darzacq X, and Tjian R (2020). Overcoming the bottleneck to widespread testing: a rapid review of nucleic acid testing approaches for COVID-19 detection. RNA 26:771-783. 10.1261/rna.076232.120
- Lu X, Wang L, Sakthivel SK, Whitaker B, Murray J, Kamili S, Lynch B, Malapati L, Burke SA, Harcourt J, Tamin A, Thornburg NJ, Villanueva JM, and Lindstrom S (2020). US CDC real-time reverse transcription PCR panel for detection of severe acute respiratory syndrome coronavirus 2. Emerg Infect Dis 26:1654-1665. 10.3201/eid2608.201246
- Nalla AK, Casto AM, Huang MLW, Perchetti GA, Sampoleo R, Shrestha L, Wei Y, Zhu H, Jerome KR, and Greninger AL (2020). Comparative performance of SARS-CoV-2 detection assays using seven different primer-probe sets and one assay kit. J Clin Microbiol 58:e00557-20. 10.1128/jcm.00557-20.
- Lu X, Whitaker B, Sakthivel SK, Kamili S, Rose LE, Lowe L, Mohareb E, Elassal EM, Al-sanouri T, Haddadin A, and Erdman DD (2014). Real-time reverse transcription-PCR assay panel for Middle East respiratory syndrome coronavirus. J Clin Microbiol 52:67-75. 10.1128/jcm.02533-13
- Leung K, Shum MH, Leung GM, Lam TT, and Wu JT (2021). Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill 26:2002106. 10.2807/1560-7917.ES.2020.26.1.2002106
- Martin DP, Weaver S, Tegally H, San JE, Shank SD, Wilkinson E, Lucaci AG, Giandhari J, Naidoo S, Pillay Y, Singh L, and Lessells RJ; NGS-SA; COVID-19 Genomics UK (COG-UK), Gupta RK, Wertheim JO, Nekturenko A, Murrell B, Harkins GW, Lemey P, MacLean OA, Robertson DL, de Oliveira T, and Kosakovsky Pond SL (2021). The emergence and ongoing convergent evolution of the SARS-CoV-2 N501Y lineages. Cell 184:5189-5200. 10.1016/j.cell.2021.09.003
- Rondinone V, Pace L, Fasanella A, Manzulli V, Parisi A, Capobianchi MR, Ostuni A, Chironna M, Caprioli E, Labonia M, Cipolletta D, Della Rovere I, Serrecchia L, Petruzzi F, Pennuzzi G, and Galante D (2021). VOC 202012/01 Variant is effectively neutralized by antibodies produced by patients infected before its diffusion in Italy. Viruses 13:276. 10.3390/v13020276
- Tegally H, Wilkinson E, Giovanetti M, Iranzadeh A, Fonseca V, Giandhari J, Doolabh D, Pillay S, San EJ, Msomi N, Mlisana K, von Gottberg A, Walaza S, Allam M, Ismail A, Mohale T, Glass AJ, Engelbrecht S, Van Zyl G, Preiser W, Petruccione F, Sigal A, Hardie D, Marais G, Hsiao NY, Korsman S, Davies MA, Tyers L, Mudau I, York D, Maslo C, Goedhals D, Abrahams S, Laguda-Akingba O, Alisoltani-Dehkordi A, Godzik A, Wibmer CK, Sewell BT, Lourenço J, Alcantara LCJ, Kosakovsky Pond SL, Weaver S, Martin D, Lessells RJ, Bhiman JN, Williamson C, and de Oliveira T (2021). Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 592:438-443. 10.1038/s41586-021-03402-9
- Tegally H, Wilkinson E, Lessells RJ, Giandhari J, Pillay S, Msomi N, Mlisana K, Bhiman JN, von Gottberg A, Walaza S, Fonseca V, Allam M, Ismail A, Glass AJ, Engelbrecht S, Van Zyl G, Preiser W, Williamson C, Petruccione F, Sigal A, Gazy I, Hardie D, Hsiao NY, Martin D, York D, Goedhals D, San EJ, Giovanetti M, Lourenço J, Alcantara LCJ, and de Oliveira T (2021). Sixteen novel lineages of SARS-CoV-2 in South Africa. Nat Med 27:440-446. 10.1038/s41591-021-01255-3
- Yuan F, Wang L, Fang Y, and Wang L (2021). Global SNP analysis of 11,183 SARS-CoV-2 strains reveals high genetic diversity. Transbound Emerg Dis 00:1-17. 10.1111/tbed.13931
- Tang X, Wu C, Li X, Song Y, Yao X, Wu X, Duan, Y, Zhang, H, Wang, Y, Qian, Z, Cui, J, and Lu, J (2020). On the origin and continuing evolution of SARS-CoV-2. Nat Sci Rev 7:1012-1023. 10.1093/nsr/nwaa036
- Starr TN, Greaney AJ, Hilton SK, Ellis D, Crawford KHD, Dingens AS, Navarro MJ, Bowen JE, Tortorici MA, Walls AC, King NP, Veesler D, and Bloom JD (2020). Deep mutational scanning of SARS-CoV-2 receptor binding domain reveals constraints on folding and ACE2 binding. Cell 182:1295-1310.e20. 10.1016/j.cell.2020.08.012
- Yi C, Sun X, Ye J, Ding L, Liu M, Yang Z, Lu X, Zhang Y, Ma L, Gu W, Qu A, Xu J, Shi Z, Ling Z, and Sun B (2020). Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol 17:621-630. 10.1038/s41423-020-0458-z
- Korber B, Fischer WM, Gnanakaran S, Yoon H, Theiler J, Abfalterer W, Hengartner N, Giorgi EE, Bhattacharya T, Foley B, Hastie KM, Parker MD, Partridge DG, Evans CM, Freeman TM, and de Silva TI; Sheffield COVID-19 Genomics Group, McDanal C, Perez LG, Tang H, Moon-Walker A, Whelan SP, LaBranche CC, Saphire EO, and Montefiori DC (2020). Tracking changes in SARS-CoV-2 Spike: evidence that D614G increases infectivity of the COVID-19 virus. Cell 182:812-827.e19. 10.1016/j.cell.2020.06.043
- Zhang L, Jackson CB, Mou H, Ojha A, Peng H, Quinlan BD, Rangarajan ES, Pan A, Vanderheiden A, Suthar MS, Li W, Izard T, Rader C, Farzan M, and Choe H (2020). SARS-CoV-2 spike-protein D614G mutation increases virion spike density and infectivity. Nature Commun 11:6013. 10.1038/s41467-020-19808-4
- Meng B, Kemp SA, Papa G, Datir R, Ferriera IATM, Marelli S, Harvey WT, Lytras S, Mohamed A, Gallo G, Thakur N, Collier DA, Mlcochova P, COVID-19 Genomics UK (COG-UK) Consortium, Duncan, LM, Carabelli AM, Kenyon JC, Lever AM, De Marco A, Saliba C, Culap K, Cameroni E, Matheson NJ, Piccoli L, Corti D, James LC, Robertson DL, Bailey D, and Gupta RK (2021). Recurrent emergence SARS-CoV-2 spike deletion H69/V70 and its role in the Alpha variant B.1.17. Cell Rep 35:109292. 10.1016/j.celrep.2021.109292
- Johnson BW, Russell BJ, and Lanciotti RS (2005). Serotype-specific detection of Dengue viruses in a fourplex real-time reverse transcriptase PCR assay. J Clin Microbiol 43:4977-4983. 10.1128/JCM.43.10.4977-4983.2005
- Lanciotti RS, Kerst AJ, Nasci RS, Godsey MS, Mitchell CJ, Savage HM, Komar N, Panella NA, Allen BC, Volpe KE, Davis BS, and Roehrig JT (2000). Rapid detection of West Nile virus from human clinical specimens, field-collected mosquitoes, and avaian samples by a TaqMan reverse transcriptase-PCR assay. J Clin Microbiol 38:4066-4071. 10.1128/JCM.38.11.4066-4071.2000
- Vijgen L, Keyaerts E, Moes E, Maes P, Duson P, and Ranst MV (2005). Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J Clin Microbiol 43:5452-5456. 10.1128/JCM.43.11.5452-5456.2005
- Kudo E, Israelow B, Vogels CBF, Lu P, Wyllie AL, Tokuyama M, Venkataraman A, Brackney DE, Ott IM, Petrone ME, Earnest R, Lapidus S, Muenker CM, Moore AJ, Casanovas-Massana A, Yale IMPACT Research Team, Omer SB, Dela Cruz CS, Farhadian SF, Ko AI, Grubaugh ND, and Iwasaki A. (2020). Detection of SARS-CoV-2 RNA by multiplex RT-qPCR. PLoS Biol 18:e3000867. 10.1371/journal.pbio.3000867
- Vogels CBF, Brito AF, Wyllie AL, Fauver JR, Ott IM, Kalinich CC, Petrone ME, Casanovas-Massana A, Muenker CM, Moore AJ, Klein J, Lu P, Lu-Culligan A, Jiang X, Kim DJ, Kudo E, Mao T, Moriyama M, Oh JE, Park A, Silva J, Song E, Takahashi T, Taura M, Tokuyama M, Venkataraman A, Weizman OE, Wong P, Yang Y, Cheemarla NR, et al. (2020). Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat Microbiol 5:1299-1305. 10.1038/s41564-020-0761-6
- Graham TGW, Dugast-Darzacq C, Dailey GM, Nguyenla XH, Van Dis E, Esbin MN, Abidi A, Stanley SA, Darzacq X, and Tjian R (2021). Open-source RNA extraction and RT-qPCR methods for SARS-CoV-2 detection. PloS One 16:e0246647. 10.1371/journal.pone.0246647
- Thul PJ and Lindskog C (2018). The human protein atlas: a spatial map of the human proteome. Protein Sci 27: 233-244. 10.1002/pro.3307
- Tani H, Mizutani R, Salam KA, Tano K, Ijiri K, Wakamatsu A, Isogai T, Suzuki Y, Akimitsu N (2012). Genome-wide determination of RNA stability reveals hundreds of short-lived noncoding transcripts in mammals. Genome Res 22: 947-956. 10.1101/gr.130559.111
- Stamova BS, Apperson M, Walker WL, Tian Y, Xu H, Adamczy P, Zhan X, Liu DZ, Ander BP, Liao IH, Gregg JP, Turner RJ, Jickling G, Lit L, and Sharp FR (2009). Identification and validation of suitable endogenous reference genes for gene expression studies in human peripheral blood. BMC Med. Genomics 2:49. 10.1186/1755-8794-2-49
- Dekker RJ, Ensink WA, van Leeuwen S, Rauwerda H, and Breit TM (2020). Overhauling a faulty control in the CDC-recommended SARS-CoV-2 RT-PCR test panel. bioRxiv. 2020:2020.06.12.147819.
- Cuevas JM, Geller R, Garijo R, López-Aldeguer J, and Sanjuán R (2015). Extremely high mutation rate of HIV-1 in vivo. PLoS Biol 13:e1002251. 10.1371/journal.pbio.1002251
- Geller R, Estada Ú, Peris JB, Andreu I, Bou JV, Garijo R, Cuevas JM, Sabariegos R, Mas A, and Sanjuán R (2016). Highly heterogeneous mutation rates in the hepatitis C virus genome. Nature Microbiol 1:16045. 10.1038/nmicrobiol.2016.45
- Lauring AS, Frydman J, and Andino R (2013). The role of mutational robustness in RNA virus evolution. Nat Rev Microbiol 11:327-336. 10.1038/nrmicro3003
- Fontanet A, Autran B, Lina B, Kieny MP, Karim SSA, and Sridhar D (2021). SARS-CoV-2 variants and ending the COVID-19 pandemic. Lancet 397:952-954. 10.1016/s0140-6736(21)00370-6
- Mascola JR, Graham BS, and Fauci AS (2021). SARS-CoV-2 viral variants-tackling a moving target. JAMA 325:1261-1262. 10.1001/jama.2021.2088
- Braasch DA and Corey DR (2001). Locked nucleic acid (LNA): fine-tuning the recognition of DNA and RNA. Chem Biol 8:1-7. 10.1016/s1074-5521(00)00058-2
- McTigue PM, Peterson RJ, and Kahn JD (2004). Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry 43:5388-405. 10.1021/bi035976d
- Petersen M and Wengel J. 2003. LNA: a versatile tool for therapeutics and genomics. Trends Biotechnol 21:74-81. 10.1016/s0167-7799(02)00038-0
- Ugozzoli LA, Latorra D, Puckett R, Arar K, and Hamby K (2004). Real-time genotyping with oligonucleotide probes containing locked nucleic acids. Anal Biochem 324:143-152. 10.1016/j.ab.2003.09.003
- Vogels CBF, Breban MI, Ott IM, Alpert T, Petrone ME, Watkins AE, Kalinich CC, Earnest R, Rothman JE, Goes de Jesus J, Morales Claro I, Magalhaes Ferreira G, Crispim MAE, Brazil-UK CADDE Genomic Network, Singh L, Tegally H, Anyaneji UJ, Network for Genomic Surveillance in South Africa, Hodcroft EB, Mason CE, Khullar G, Metti J, Dudley JT, MacKay MJ, Nash M, Wang J, Liu C, Hui P, Murphy S et al. (2021). Multiplex qPCR discriminates variants of concern to enhance global surveillance of SARS-CoV-2. PLoS Biol 19:e3001236. 10.1371/journal.pbio.3001236
- Bustin S, Coward A, Sadler G, Teare L, and Nolan T (2020). CoV2-ID, a MIQE-compliant sub-20-min 5-plex RT-PCR assay targeting SARS-CoV-2 for the diagnosis of COVID-19. Sci Rep 10:22214. 10.1038/s41598-020-79233-x
- Roumani F, Azinheiro S, Sousa H, Sousa A, Timoteo M, Varandas T, Fonseca-Silva D, Baldaque I, Carvalho J, Prado M, and Garrido-Maestu A (2021). Optimization and clinical evaluation of a multi-target loop-mediated isothermal amplification assay for the detection of SARS-CoV-2 in nasopharyngeal samples. Viruses 13:940. 10.3390/v13050940
- Kidd M, Richter A, Best A, Cumley N, Mirza J, Percival B, Mayhew M, Megram O, Ashford F, White T, Moles-Garcia E, Crawford L, Bosworth A, Atabani SF, Plant T, and McNally A (2021). S-variant SARS-CoV-2 lineage B1.1.7 is associated with significantly higher viral loads in samples tested by ThermoFisher TaqPath RT-qPCR. J Infect Dis 223:1666-1670. 10.1093/infdis/jiab082
- Leung K, Shum MH, Leung GM, Lam TT, and Wu JT (2021). Early transmissibility assessment of the N501Y mutant strains of SARS-CoV-2 in the United Kingdom, October to November 2020. Euro Surveill 26:2002106. 10.2807/1560-7917.ES.2020.26.1.2002106
- Longtin Y, Charest H, Quach C, Savard P, Baz M, Boivin G, Farfard J, Villeneuve J, Roger M, and De Serres G (2021). Infectivity of healthcare workers diagnosed with coronavirus disease 2019 (COVID-19) approximately 2 weeks after onset of symptoms: A cross-sectional study. Infect Control Hosp Epidemiol 1-3. 10.1017/ice.2020.1420
- Ginzinger DG (2002). Gene quantification using real-time quantitative PCR: an emerging technology hits the mainstream. Exp Hematol 30:503-12. 10.1016/s0301-472x(02)00806-8
- Vijgen L, Keyaerts E, Moës E, Maes P, Duson G, and Van Ranst M (2005). Development of one-step, real-time, quantitative reverse transcriptase PCR assays for absolute quantitation of human coronaviruses OC43 and 229E. J Clin Microbiol 43:5452-5456. 10.1128/jcm.43.11.5452-5456.2005
- Fronhoffs S, Totzke G, Stier S, Wernert N, Rothe M, Brüning T, Koch B, Sachinidis A, Vetter H, and Ko Y (2002). A method for the rapid construction of cRNA standard curves in quantitative real-time reverse transcription polymerase chain reaction. Mol Cell Probes 16:99-110. 10.1006/mcpr.2002.0405
- Poritz MA, Blaschke AJ, Byington CL, Meyers L, Nilsson K, Jones DE, Thatcher SA, Robbins T, Lingenfelter B, Amiott E, Herbener A, Daly J, Dobrowolski SF, Teng DH, and Ririe KM (2011). FilmArray, an automated nested multiplex PCR system for multi-pathogen detection: development and application to respiratory tract infection. PloS One 6:e26047. 10.1371/journal.pone.0026047
–
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
AUTHOR CONTRIBUTIONS
M.D. conceptualized, performed and analyzed rRT-PCR data. P.T. conceptualized the design of primers and TaqMan-LNA probes as well as carrying nu-merous bioinformatic analyses. S. Lévesque conceptual-ized and performed BioFire FilmArray assays. A.B. ac-quired and analyzed the data. A.C, L.V., and P.M. concep-tualized the experimental work and analyzed data. S. Labbé conceptualized the experimental work and wrote the manuscript. The authors reviewed the results and approved the final version of the manuscript.
ACKNOWLEDGMENTS
We are grateful to Dr. Gilles Dupuis for critical reading of the manuscript and for his valuable comments. We thank Dr. Hugues Charest for his com-munication with respect to the N LSPQ primer-probe set and its utilization in rRT-PCR assays.
COPYRIGHT
© 2021

Detection of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and its first variants in fourplex real-time quantitative reverse transcription-PCR assays by Durand et al. is licensed under a Creative Commons Attribution 4.0 International License.








