
Research Articles:
Microbial Cell, Vol. 3, No. 2, pp. 53 - 64; doi: 10.15698/mic2016.02.476
Inhibition of Aβ42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs
1 Present address: Department of Biochemistry and Molecular Biology, University of Nevada, Reno, Reno, NV, USA.
2 HTS facility, Research Resources Center, University of Illinois, Chicago, Chicago, IL 60612, USA.
3 Department of Biological Sciences, University of Illinois, Chicago, Chicago, IL 60607, USA.
Keywords: Aβ42 oligomerization, yeast, HTS, PICALM, Alzheimer.
Abbreviations:
AD - Alzheimer disease,
Aβ - beta-amyloid,
FDA - Food and Drug Administration agency,
HTS - high-throughput screening,
PGK - 3-phosphoglycerate kinase,
PICALM - phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia,
RF - release factor,
TMR - tetramethylrhodamine.
Received originally: 01/08/2015 Received in revised form: 03/12/2015
Accepted: 08/12/2015
Published: 20/01/2016
Correspondence:
Susan W. Liebman, Department of Biochemistry and Molecular Biology, University of Nevada, Reno 1664 N. Virginia Street, Mail stop/330; Reno NV89557, USA sueL@uic.edu
Conflict of interest statement: The authors declare there is no conflict of interest.
Please cite this article as: Sei-Kyoung Park, Kiira Ratia, Mariam Ba, Maria Valencik and Susan W. Liebman (2016). Inhibition of Aβ42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs. Microbial Cell 3(2): 53-64.
Abstract
The formation of small Aβ42 oligomers has been implicated as a toxic species in Alzheimer disease (AD). In strong support of this hypothesis we found that overexpression of Yap1802, the yeast ortholog of the human AD risk factor, phosphatidylinositol binding clathrin assembly protein (PICALM), reduced oligomerization of Aβ42 fused to a reporter in yeast. Thus we used the Aβ42-reporter system to identify drugs that could be developed into therapies that prevent or arrest AD. From a screen of 1,200 FDA approved drugs and drug-like small compounds we identified 7 drugs that reduce Aβ42 oligomerization in yeast: 3 antipsychotics (bromperidol, haloperidol and azaperone), 2 anesthetics (pramoxine HCl and dyclonine HCl), tamoxifen citrate, and minocycline HCl. Also, all 7 drugs caused Aβ42 to be less toxic to PC12 cells and to relieve toxicity of another yeast AD model in which Aβ42 aggregates targeted to the secretory pathway are toxic. Our results identify drugs that inhibit Aβ42 oligomers from forming in yeast. It remains to be determined if these drugs inhibit Aβ42 oligomerization in mammals and could be developed as a therapeutic treatment for AD.
INTRODUCTION
Alzheimer’s disease (AD), a progressive and fatal brain disorder, is the most common form of dementia currently affecting more than 5 million Americans. Furthermore, more than 26 million people worldwide have some form of dementia.
–
With the increase in the age of the population, the number of AD patients is expected to triple by 2050 causing a staggering emotional and financial toll. Unfortunately, there is no prevention or satisfactory treatment to date.
–
To prevent or stop disease progression at an early stage, we need to attack the underlying causes of the disease. The major physical feature of AD is the accumulation of abnormally folded beta-amyloid (Aβ) and Tau in the brain. When Aβ42 is polymerized in vitro [1][2][3][4] or purified from in vivo animal models, post mortem brain [5][6] or cerebrospinal fluid (CSF) tissue of AD patients [7][8], small Aβ42 aggregates are found that differ in size and shape. Such small aggregates of the Aβ42 peptide (dimers, trimers, tetramers, etc.) appear to be neurotoxic because they trigger abnormalities in neuronal excitation and synaptic plasticity, and inhibit hippocampal long-term potentiation [1][2][9][10][11][12]. Still, it remains to be determined if Aβ42 is a major cause of AD [13]. Nonetheless, there is evidence for a pathological role of Aβ oligomers on other protein oligomers in neurodegenerative conditions, such as Parkinson’s disease [14], and prion diseases [15].
–
Many fundamental biological processes and pathways such as chaperone and protein remodeling, the ubiquitin proteasome system, secretion, vesicular trafficking, and autophagy, are highly conserved between yeast and human cells. Indeed, yeast models have become powerful tools for unraveling the molecular basis of complex human neurodegenerative diseases [16][17][18][19]. Treatment with Aβ42 oligomers formed in vitro or expression of Aβ42 oligomers in vivo affects the growth of yeast cells [20][21][22][23][24]. Also, a yeast model in which Aβ42 (referred as HDEL-Aβ42) is toxic was recently developed. Here, a GAL1 promoter was used to express a KAR2 signal sequence (HDEL) Aβ42 fusion protein. This Aβ42 fusion protein was directed to the secretory pathway where it disrupted normal cellular endocytic trafficking, causing toxicity [24]. Importantly, overexpression of YAP1802, a yeast homolog of PICALM, rescued cells from this toxicity [24]. PICALM (phosphatidylinositol clathrin assembly lymphoid-myeloid leukemia) protein plays a key role in a clathrin-mediated endocytosis and genome-wide association studies identified single nucleotide polymorphisms in the gene of PICALM as genetic risk factors for late-onset AD [25][26]. Furthermore, overexpressed PICALM protected primary rat cortical neurons and C. elegans from toxicity of extracellular aggregated Aβ42 oligomers. In addition, PICALM affected Aβ42 toxicity in a yeast model in which the α-factor signal sequence was fused to Aβ42, although here overexpression of PICALM enhanced toxicity [23]. While little is known about the contribution of PICALM to AD pathogenesis, these findings strongly support the hypothesis that Aβ42 is associated with AD toxicity.
–
Yeast has been used to screen for chemical compounds that reduce aggregation or oligomerization of the Aβ peptide by assaying for the activity of reporters fused to Aβ [27][28]. Previously, using a yeast Aβ42 oligomerization model in which Aβ42 was fused to the functional release factor (RF) domain of yeast translational termination factor, Sup35 [29], we screened for anti-Aβ42 oligomer compounds [28]. This Aβ42-RF fusion formed SDS-resistant low-n oligomers that reduced release factor activity, thus enhancing a read-through of stop codon mutations. Indeed, a correlation of oligomer formation and stop codon read-through was confirmed by biochemical analysis [28][29]. An important distinction of this approach from previous anti-Aβ aggregation screens [30][31][32] is that we can detect drugs that inhibit Aβ42 oligomer formation but do not inhibit the formation of large Aβ42 amyloid. This is important because such large aggregates are now thought to be helpful because they likely capture some of the more toxic Aβ42 oligomers, rendering them less toxic [33][34]
–
Here, we show that the mechanism of the PICALM, human AD risk factor, is likely to reduce the level of Aβ42 oligomers in cells. This strongly supports the hypothesis that oligomerization of Aβ42 is a major cause of AD toxicity. We then screened FDA-approved drugs that could readily be developed into Alzheimer’s therapies, to identify drugs that prevent the formation of Aβ42 small oligomers using the yeast Aβ42-RF reporter system. We also showed that each of the drug hits counteract yeast and mammalian cell toxicity associated with Aβ42 small aggregates.
RESULTS
YAP1802, homolog of PICALM, inhibits Aβ42-RF oligomer formation
We tested whether genetic modifiers that rescue HDEL-Aβ42 toxicity [24] likewise repair the compromised Aβ42-RF translational termination factor activity due to reduced Aβ42-RF oligomer formation using the above described growth assay [28][29]. The cell growth phenotype in this assay requires expression of full length Ade1. The impaired Sup35 translational release factor (RF) activity of oligomerized Aβ42-RF allows read-through of the ade1-14 premature stop codon enabling growth on adenineless media (-Ade) (Aβ42-RF-empty vector (e. v.) in Figure 1A). However in the presence of drugs or genetic modifiers blocking oligomerization of Aβ42-RF, or in cells expressing a fusion made with the Aβ42 aggregation-deficient mutation [28][29], Aβ42m2-RF, the RF activity is restored, so cells cannot grow on -Ade (Aβ42m2-RF-empty vector (e. v.) in Figure 1A). Among the 12 genetic modifiers identified in the HDEL-Aβ42 screen [24], only cells expressing YAP1802 regained translational termination factor activity (shown as reduced growth on -Ade), suggesting it restored Aβ42-RF to the soluble monomeric state (Figure 1A). Indeed, immunoblots developed with Sup35C antibody showed that the level of SDS-resistant Aβ42-RF oligomer was significantly reduced, and the level of the monomers was significantly increased when Yap1802 was overexpressed (Figure 1B). Thus, YAP1802 is likely to affect toxicity by reducing the level of toxic Aβ42 oligomers.
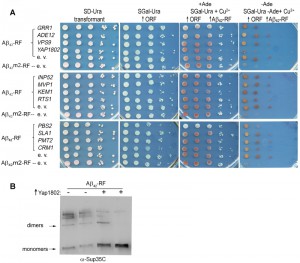 | FIGURE 1: Effect of genetic modifiers of toxic HDEL-Aβ42 on Aβ42-RF oligomer formation and Aβ42-MRF aggregates into toxic SDS-stable oligomers in yeast. (A) Overexpressed Yap1802 but not other previously described [24] HDEL-Aβ42 modifiers dramatically enhanced the translational termination factor activity of Aβ42-RF. Aβ42-RF expressing cells (CUP1::Aβ42-RF) lacking chromosomal SUP35 were transformed with a plasmid carrying the indicated gene under control of the GAL1 promoter. Ten-fold serial dilutions of transformants are shown on plasmid selective non-inducing media (SD-Ura); galactose plasmid selective media (SGal-Ura) to turn on each gene (↑ORF); +Ade medium containing copper to overexpress Aβ42-RF (↑ORF ↑Aβ42-RF); the identical -Ade medium SGal-Ura-Ade+Cu2+ (-Ade ↑ORF ↑Aβ42-RF) to determine translational termination factor activity as read-through of the ade1-14 nonsense mutant. The Aβ42-RF overexpressed in cells was shown to have reduced translational termination factor activity as cells grew on -Ade due to aggregation of the fusion protein into small oligomers (Aβ42-RF-e. v.). However, the translation termination factor activity was retained in yeast cells overexpressing Aβ42m2-RF (Aβ42 aggregation-deficient mutant) or YAP1802, due to the absence of oligomer formation, resulting in reduced growth on -Ade (Aβ42m2-RF-e. v.). (B) Yap1802 suppression of Aβ42-RF oligomerization by immunoblot analysis. Total cell lysates were prepared from 2 independent transformants of an Aβ42-RF strain carrying YAP1802 (+) or an empty vector (-) plasmid. Both Aβ42-RF and YAP1802 were overexpressed (SGal-Ura-Ade+Cu2+) prior to lysis. Immunoblots were probed with anti-Sup35 RF to evaluate the level of oligomers and monomers. ↑Yap1802 indicates overexpression of YAP1802. The identification of bands as Aβ42-RF dimers and monomers was determined by the estimated sizes of the bands in the immunoblot. |
We also used NAB61 antibody, which was reported to preferentially recognize toxic Aβ oligomers in AD patients [35] and HDEL-Aβ42 in yeast [24]. Indeed, while Sup35C antibody detected only monomers in Aβ42m2-RF cells expressing the Aβ42 aggregation-deficient mutation (Figure S1), NAB61 did not recognize Aβ42m2-RF monomers although it detected a species the size of dimers. However, in our hands NAB61 recognized both oligomers and monomers of wild-type Aβ42-RF (Figure S1).
–
Screen for drugs that inhibit Aβ42-RF from forming toxic oligomers
We screened the Prestwick Chemical Library® (Prestwick Chemical, Washington, DC) which contains 1,200 FDA approved drugs and drug-like molecules for the ability to reduce Aβ42 oligomer formation using the growth assay described above [28][29]. Drugs that blocked oligomerization of Aβ42-RF, thereby restoring the RF activity and preventing stop codon read-through, were selected as initial candidates because they reduced growth in -Ade.
–
Each assay was carried out in duplicate at a single compound concentration (20 µM). The Z values [36] calculated from controls were 0.65 and 0.71 for the -Ade and +Ade assays, respectively, indicating the assay qualities were excellent. In the initial screen, 24 known drugs from among the 1,200 compounds in the library emerged as candidates that inhibited growth in -Ade medium without significant inhibition of growth in +Ade medium. When the results were repeated, 14 of the 24 drugs at 20 µM in DMSO inhibited growth more than 50% in -Ade when compared to a no drug DMSO control (the 8 drugs in Figure 2 and the 6 drugs written in bold in Figure S2). The 6 hits shown in Figure S2 were dropped because they also decreased growth in +Ade medium by ≥30% compared to the no drug control, indicating they are generally toxic to yeast, which invalidates our -Ade growth assay.
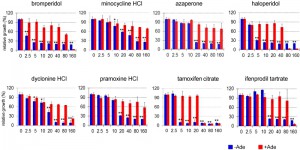 | FIGURE 2: Dose-dependent effects of 8 drugs on Aβ42-RF activity as measured by cell growth. Aβ42-RF expressing cells were treated with each drug at the indicated concentrations. Their effects on cell growth were measured as OD600 in +Ade and -Ade media after 4 and 5 days respectively (see Materials and Methods for details). Growth in -Ade is dependent upon Aβ42-RF release factor activity. This activity is expected to be reduced by oligomerizartion. Growth on +Ade does not require Aβ42-RF release factor activity and is used as a control in the presence of each drug. Shown is the relative growth in the presence vs. absence of each drug. Error bars show the standard deviation from three replicates. The asterisks show significance levels of **P < 0.01 or *P < 0.05 according to the Student’s t test between DMSO (0) control and drug treatment at marked concentration for growth in -Ade. |
Dose-dependent inhibition of Aβ42-RF oligomerization
The 8 drugs that passed the confirmation screen were tested for dose-dependent effects on Aβ42-RF activity. The percent of relative growth in the presence of the drugs dissolved in DMSO was determined from the OD600 (Figure 2). The Aβ42-RF control treated with DMSO only, showed good growth in -Ade, indicating Aβ-RF associated translational termination activity is compromised in the absence of drug due to oligomerization of the fusion protein. Aβ42m2-RF with DMSO had little or no growth in -Ade, indicating the mutant fusion protein was functional and not aggregated (Figures 1A and S1). In the presence of each drug, Aβ42-RF showed dose-dependent growth inhibition in -Ade, but no or mild growth inhibition in +Ade, indicating that the drugs block Aβ42-RF activity likely by blocking oligomerization.
–
We analyzed lysates prepared from cells grown with various concentrations (0-40 or 0-160 µM) of each drug to test whether the restored translational termination factor activity associated with the drugs is correlated with a decrease in the level of SDS-resistant Aβ42-RF oligomers. In cells grown with DMSO only (the first lane marked 0 in each gel in immunoblots in Figure 3), there were mostly SDS-resistant Aβ42-RF oligomers (dimers, trimers and tetramers, etc.) and few monomers. Higher drug concentrations decreased the levels of oligomers and concomitantly increased the level of monomers, while the drugs have no effect on 3-phosphoglycerate kinase (PGK) expression at any concentration. This inhibition of oligomer formation was quantified by comparing the ratios of oligomers to monomers detected on immunoblots of cell lysates grown in the presence or absence of drug (Figure 3 shows a representative sample from 3 independent experiments). Bromperidol, azaperone, haloperidol, pramoxine HCl, and dyclonine HCl exhibited strong anti-oligomer activity (Figure 3) as well as restored Aβ42-RF activity indicated by growth inhibition in -Ade at relatively low concentrations (Figure 2). Unexpectedly, minocycline HCl and tamoxifen citrate, that decreased growth in -Ade at 10-20 µM and 5 µM, respectively (Figure 2), only showed inhibition of oligomer formation at high concentrations, 160 and 80 µM, respectively. Finally, ifenprodil tartrate that showed dose-dependent growth inhibition in -Ade failed to change the oligomer profile (Figure 3) and was dropped.
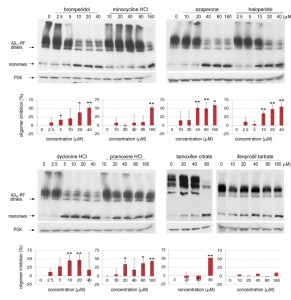 | FIGURE 3: Seven drugs suppress Aβ42-RF oligomerization in yeast in a dose-dependent manner. SDS-resistant Aβ42-RF oligomers were detected by immunoblot analysis (upper). The assay strain expressing Aβ42-RF was grown in complex medium in the presence of each compound at the indicated concentrations. Equal amounts of lysate proteins were treated with 1% SDS for 7 mins at room temperature and analyzed by SDS PAGE followed by immunoblotting with anti-Sup35 RF antibodies. Aβ42-RF cells grown in DMSO (0) were used as controls. PGK, yeast 3-Phosphoglycerate Kinase, detected with anti PGK antibodies was an internal control to show the effect of the drugs on protein synthesis in general. Immunoblot signals for Aβ42-RF monomers and oligomers were quantified and converted into % inhibition of oligomer formation from the ratios of oligomers to monomers compared to DMSO controls (0 means no inhibition) (lower). Error bars represent standard deviation from 3 independent immunoblots except for ifenprodil tartrate which shows data from 1 of 2 immunoblots that showed no drug effects. The asterisks show significant levels of **P < 0.01 or *P < 0.05 according to Student’s t test. |
Importantly, the remaining 7 drugs enhanced the ability of Aβ42-RF, but not of a SUP35 suppressor mutation (G1256A [37]), to terminate protein synthesis at the ade1-14 nonsense mutation, as measured by growth on -Ade (Figure S3). Thus the drugs do not have a general antisuppressor activity, but instead specifically affect Aβ42-RF.
–
Drugs prevent Aβ42 from being toxic in PC12 cells
Some species of Aβ oligomers of various sizes and shapes prepared in vitro or purified from post mortem brains have been shown to be toxic when applied to neuronal cell culture or primary cortical neurons [38][39][40][41]. To test the activity of drugs in reducing Aβ42 oligomerization, we added the drugs as we assembled Aβ42 using in vitro conditions in which the aggregation of Aβ42 into small soluble oligomers was favored [38]. Treatment of rat PC12 neuronal cells with 20 µM of Aβ42 assembled in the presence of DMSO decreased cell viability to about 40% (red bar in Figure 4A) compared to control cells treated with DMSO in the absence of Aβ42 (blue bar in Figure 4A), indicating that the assembled Aβ42 were indeed toxic. Aβ42 assembled in the presence of the remaining 7 hits from the Prestwick Chemical Library® were less toxic (Figure 4A), suggesting that the drugs impede the formation of the toxic Aβ42 assemblies. In addition, AO-11 and AO15, compounds that inhibit Aβ42-RF from forming oligomers, identified from our previous screen [28], also relieved toxicity.
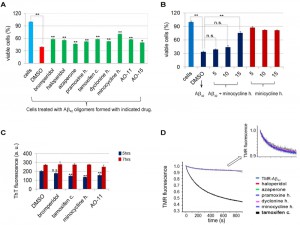 | FIGURE 4: The protective effect of drugs on Aβ42 induced cytotoxicity in PC12 cells. (A) Toxicity of Aβ42 assembled under conditions favoring oligomer formation was reduced when assembly was done in the presence of drugs. Aβ42 at 200 μM assembled under conditions favoring oligomer formation in the presence of 100 μM drug or DMSO was diluted and added to PC12 cells at a concentration of 20 μM Aβ42 and 10 μM drug. Cell viability was assessed after 24 hrs growth at 37°C using the MTT assay. The percentage of viable cells is shown. Error bars are the standard deviation of triplicate experiments. (B) Dose-dependent effects of minocycline HCl on Aβ42 associated cytotoxicity. Methods were as in (A). PC12 cells were treated with final concentrations of 20 μM Aβ42 with 5, 10, and 15 µM minocycline HCl or DMSO only (blue bars). Cells were also treated with the indicated amount of minocycline HCl alone (red bars). (C) Effect of drugs on in vitro Aβ42 high molecular fibril assembly in fibril favorable conditions. Aβ42 was assembled into high molecular fibrils at 37°C in the presence of 50 µM drug or control DMSO according to [42]. Thioflavine T (ThT) was added to aliquots at the indicated times and fluorescence, indicative of fiber formation, was measured. (D) Effect of drugs on in vitro labeled Aβ42 (TMR-K-Aβ42) oligomerization. Reduced TMR fluorescence, which measures oligomerization, was recorded as a function of time immediately after dilution of the TMR-K-Aβ42 with addition of 10 µM of each drug or DMSO for a negative control. The asterisks show significance levels of **P < 0.01 or *P < 0.05 according to Student’s t test between DMSO control and drug treatment (A) and DMSO control and drug treatment at 5 hrs (C). |
Among drugs tested, minocycline HCl was the most effective. Cell viability increased from 40% when Aβ42 was assembled in DMSO alone, to 68% when it was assembled in the presence of 10 µM minocycline HCl (Figure 4A). The minocycline HCl protection was dose-dependent (Figure 4B). Furthermore, treatment of PC12 cells with minocycline HCl without Aβ42 did not improve cell viability (Figure 4B).This suggests the protective effect of the minocycline HCl was not due to the enhancement of cell growth but rather was due to inhibition of Aβ42 toxicity.
–
Drugs do not affect in vitro Aβ42 fibril or oligomer formation
To test whether the drugs can also inhibit high molecular Aβ42 fibril formation, 4 of the most active drugs (bromperidol, tamoxifen citrate, minocycline HCl, and AO-11) were added to 20 µM Aβ42 at 50 µM, under conditions that promote fibrilization [42]. Fibrils were allowed to form for 5 or 72 hrs and the Thioflavine T (ThT) fluorescence assay was used to quantify fibril formation. At 5 hrs there was a mild inhibition of Aβ42 fibrilization, but by 72 hrs there was no significant change in fibrilization relative to the DMSO control (Figure 4C). Thus, while these drugs may have some effect on initial Aβ42 small seed formation, they do not block fibril formation.
–
We also tried to test the effects of bromperidol, azaperone, pramoxine HCl, dyclonine HCl, minocycline HCl and tamoxifen citrate when added to Aβ42 during in vitro oligomerization conditions [38]. However, when these reactions were run on Western blots, only Aβ42 the size of 12mers and larger were detected. Aβ42 monomers and small oligomers were not seen and the drugs had no effect on this.
–
Tamoxifen citrate prevents early steps of Aβ42 oligomerization in vitro
We used an in vitro labeled Aβ42 oligomerization assay to test the effects of the drugs on the early steps of monomer to oligomer conversion [43]. This assay takes advantage of the fluorescence self-quenching observed when tetramethylrhodamine (TMR) is covalently attached to the N-terminal lysine of Aβ42 (TMR-K-Aβ42) [43]. The loss of TMR fluorescence indicates self-association of Aβ42 monomers into oligomers (e.g. dimers, trimers, etc.). Most of the drugs did not affect the formation of the early oligomers in this assay. They showed the same fluorescence changes as the control without drug (TMR-K-Aβ42) (Figure 4D). In contrast, tamoxifen citrate strongly reduced TMR fluorescence, indicating the formation of early off-pathway oligomers [43] that could be functional or non-functional oligomers.
–
Anti-Aβ42-RF oligomer drugs reduce toxicity of HDEL-Aβ42 in yeast
We next tested the effects of the 7 drugs we identified that impede Aβ42-RF oligomerization (Figures 2 and 3) on HDEL-Aβ42 toxicity. To do this we used a strain with a deletion of ERG6 (L3340), which is critical for increased drug permeability. L3340 was transformed with a 2µ plasmid (pAG425 GAL1::HDEL-Aβ42) carrying HDEL-Aβ42 under the GAL1 promoter. As previously reported for ERG6 wild-type cells [24], we found that erg6∆ cells expressing HDEL-Aβ42 were growth inhibited (middle in Figure S4) compared to controls transformed with an empty vector (top in Figure S4) and that the toxicity was suppressed by overexpressing YAP1802 (bottom in Figure S4).
–
We measured growth in liquid to assay the effects of drugs that inhibit Aβ42-RF oligomerization on the toxicity of this strain (Figure 5). Drugs at the indicated concentrations were added to yeast with the toxic HDEL-Aβ42 construct or an empty vector control. Cells were diluted in media that did (SGAL-Leu) or did not (SR-Leu) induce expression of HDEL-Aβ42. While controls carrying the empty vector grew well in both inducing and non-inducing media (dark green bars in Figure 5), growth of yeast induced to express the HDEL-Aβ42 (black bar in Figure 5 left) was decreased by 65% compared to growth in non-inducing media (black bar in right), verifying that HDEL-Aβ42 caused cytotoxicity. Treatment with 10 and 20 µM bromperidol, haloperidol and minocycline HCl relieved the toxicity while higher drug concentrations were toxic. Treatment with azaperone, pramoxine HCl, tamoxifen citrate and dyclonine HCl showed a relatively strong effect of suppressing the toxicity HDEL-Aβ42 (left panel Figure 5) while not having a significant effect on growth in the absence of HDEL-Aβ42 (right panel Figure 5). However, we were unable to detect effects of drugs tested (azaperone, pramoxine HCl, dyclonine HCl and tamoxifen citrate) on the HDEL-Aβ42 oligomerization. Possibly this is because oligomerization of HDEL-Aβ42 was very different from the oligomerization of Aβ42-RF, showing only HDEL-Aβ42 monomers and trimers.
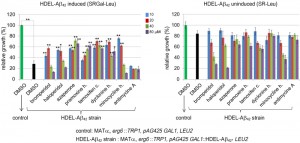 | FIGURE 5: The effect of drugs that inhibit Aβ42-RF from forming oligomers on toxicity caused by overexpression of HDEL-Aβ42 in yeast. Effects of 7 drugs on suppression of HDEL-Aβ42 toxicity and general cell growth are shown. Yeast cells carrying either HDEL-Aβ42 (HDEL-Aβ42 strain) or an empty vector (control) grown overnight in non-inducing plasmid selective media (2% raffinose, SR-Leu) were normalized, diluted to 1 x 105 cells/100 µl of media in each of 96 wells and further grown for 3 days in 2% galactose inducing media (SRGAL-Leu) or 2% raffinose noninducing media (SR-Leu) in the presence of each drug at the indicated concentrations. Growth was measured as OD600. Error bars show the standard deviation from three trials. The asterisks show increased relative growth with drug treatment compared to DMSO control (black bar in left panel) at significance levels of **P < 0.01 or *P < 0.05 according to Student’s t test. |
Antimycine A, a control that showed no effect on Aβ42-RF oligomerization (Figure S5) was also unable to relieve toxicity of HDEL-Aβ42. In addition, the other 15 drugs that initially passed the Aβ42-RF growth inhibition screen, but that were later eliminated as candidates, all failed to rescue HDEL-Aβ42.
DISCUSSION
Despite tremendous efforts, there are only a few drugs that mask or relieve the symptoms of AD. Furthermore, we do not have any medications that prevent, arrest or cure the disease (www.alz.org). Recent improved assays provide evidence for Aβ small oligomeric species in AD brains [7][8][44]. While the cause of AD remains unknown, increasing evidence suggests that aggregation of the Aβ42 peptide into small oligomers plays an important role in disease progression [12][15][45]. This hypothesis is also supported by a yeast model [24] in which toxicity associated with expression of Aβ42 was suppressed by overexpression of YAP1802, the yeast homolog of PICALM, a previously known genetic AD risk factor [25][46]. Furthermore, in this paper we show that overexpressing Yap1802 inhibits Aβ42-RF aggregation into oligomeric species (Figure 1B). This provides strong support for the idea that inhibiting the initial production or aggregation of Aβ42 would be an effective treatment to prevent or slow disease onset.
–
One approach has been to prevent the production of Aβ peptide by inhibiting the secretases that cleave Aβ from the amyloid precursor protein [47][48]. However, results of recent clinical trials of inhibitors of β- (LY2886721; Eli Lilly and Company, 2013 press release) and γ-secretases (Semagacestat; Eli Lilly and Company, 2010 press release and Avagacestat; Bristol-Myers Squibb, 2012 press release) have been disappointing. This makes the approach of inhibiting the formation of toxic Aβ oligomers of more interest.
–
Among a handful of drugs that appear to inhibit Aβ42 from forming toxic oligomers [6][27][32][41][49][50][51][52][53] in pilot studies, several have reached clinical trials. Indeed, trials of PBT1 (clioquinol), a metal chelator, which modulates affinity for Cu2+ and Zn2+ and inhibits metal-induced Aβ42 aggregation [6][54][55] were dropped because of side effects [56]. PBT2, a second generation of PBT1, was safe but demonstrated no significant effect on cognition or memory in the trial (Pubmed Health, 2014). Phase 2 trials of scyllo-inositol (ELND005), that targets the C-terminus of Aβ42 and neutralizes cell derived Aβ42 trimers [57], were completed with promising results (Transition Therapeutics, Inc., 2014 news press).
–
Finding good drug candidates through high-throughput screening (HTS) is a long and costly process and is often challenged by lack of an appropriate system, low hit rates with high false-positives and toxicity of compounds. In this study, we use a yeast cell-based HTS to identify compounds that inhibit the Aβ42-RF oligomerization among a library composed of FDA approved marketed drugs or drug-like compounds. Eight drug candidates emerged from the HTS screen (Figure 2), and 7 were found to have anti-Aβ42 oligomeric properties in subsequent biochemical assays (Figure 3): 3 antipsychotics (bromperidol, haloperidol and azaperone), 2 anesthetics (pramoxine HCl and dyclonine HCl), tamoxifen citrate, and minocycline HCl. Furthermore, the anti-oligomeric activity in growing yeast, of each of these 7 drugs, increased with increased dosages. Among our 7 hits, bromperidol, haloperidol, and azaperone are in the same drug class: dopamine antagonists. Pramoxine HCl and dyclonine HCl are both anesthetics.
–
Since the drugs we identified are already used in humans to treat other diseases, their side effects are known to be tolerable. In addition, at least four of these drugs – bromperidol, haloperidol, azaperone, and minocycline HCl, pass the blood brain barrier. These are both hurdles that eliminate most Alzheimer’s drug candidates from further study. Thus epidemiological studies of Alzheimer incidence in patients that take these drugs is now warranted.
–
Importantly, we linked the Aβ42-RF oligomers formed in yeast with a possible pathological toxic oligomer species of the HDEL-Aβ42 fusion that was directed to and disrupted normal cellular endocytic trafficking in yeast [24]. While Yap1802 restored endocytic trafficking function to signaling molecules perturbed by Aβ42 aggregates [24], and PICALM protected rat cortical neurons from toxicity of Aβ42 oligomers formed extracellularly [24], it was not clear how PICALM actually impacted Aβ toxicity. Here, by testing genetic modifiers of HDEL-Aβ42 toxicity for those that repair the compromised Aβ42-RF translational termination factor activity we found that cells overexpressing YAP1802 regained translational termination factor activity, suggesting it restored Aβ42-RF to the soluble monomeric state (Figure 1A). Indeed, overexpressed Yap1802 reduced the level of SDS-resistant Aβ42-RF oligomer by more than 50% (Figure 1B), indicating that Yap1802 is likely to have a common role in aggregation and toxicity of Aβ42.
–
As expected, the other anti-Aβ42-RF oligomer drugs also rescued toxicity of HDEL-Aβ42 (Figure 5). In contrast, the other 11 modifiers of HDEL-Aβ42 activity (shown in Figure 1A) did not have any effects on the Aβ42-RF system. This is not unexpected as these modifiers may affect secretion rather than oligomer formation of the peptide. Also all of the 7 anti-Aβ42-RF oligomerization drugs had effects on toxicity of Aβ42 rat PC12 cells (Figure 4A) and another toxic yeast AD system (Figure 5). This validates the Aβ42-RF system as a useful screen for Alzheimer’s drugs.
–
In our study the relative effectiveness of each drug was not always consistent in different assays, leaving the most relevant assay to be determined. Minocycline HCl and tamoxifen citrate only suppressed Aβ42-RF oligomerization at high concentrations (Figure 3), but had among the strongest effects on oligomer levels and toxicity (Figures 2-5). Furthermore, only tamoxifen citrate inhibited in vitro oligomerization of TMR-Aβ42 (Figure 4D), indicating that minocycline HCl and tamoxifen citrate may inhibit Aβ42 toxicity via distinct pathways.
–
It is interesting that minocycline HCl and tamoxifen citrate, which were identified as strong hits in this study, are already being used in clinical trials for other neurodegenerative diseases, such as Amyotrophic Lateral Sclerosis and Huntington Disease (https://clinicaltrials.gov/). Furthermore, minocycline HCl is a semi-synthetic tetracycline antibiotic that effectively crosses the blood-brain barrier. It has been suggested that minocycline protects patients from brain damage by reducing inflammation in AD, related tauopathies and neurodegenerative diseases that are caused by misfolded proteins [58][59][60][61]. Tamoxifen is an antagonist of calmodulin, a major cellular calcium receptor and calcium dependent regulator of many cellular processes. Increased calcium level alters neuronal dysfunction and ultimately leads to cell death in AD brains, showing there is a connection between calcium and AD. We now suggest that minocycline HCl and tamoxifen citrate also have anti-Aβ42 oligomeric activity.
–
The recent detection of potential hybrid oligomers composed of Aβ and other neurodegenerative disease associated proteins such as α-synuclein, TDP-43 and PrP from human AD post mortem brains suggest that Aβ oligomers may act as a template for the aggregation of other proteins generating a secondary amyloidosis [14][15][62]. Thus, prevention of Aβ peptide aggregation into toxic oligomers could not only prevent AD, but might also remove oligomeric seeds that contribute to the progression of other neurodegenerative diseases.
–
Our results demonstrate the efficacy of the HTS screen for drugs that inhibit Aβ42-RF oligomer formation. Future determination of the cellular targets of the drug hits may aid drug discovery. Most importantly it remains to be determined if these drugs can inhibit Aβ42 from forming toxic oligomers in humans, thereby reducing Alzheimer symptoms.
MATERIALS AND METHODS
Yeast strains, media and plasmids
L3149 and L3150 [28], used to test the effects of drugs on Aβ42-RF oligomerization, are SUP35 and ERG6 disrupted versions of 74-D694 (MATa ade1-14 ura3-52 his3-200 sup35∆::LEU2 erg6∆::TRP1), respectively containing p1364 (pRS313, CEN, URA3, CUP1::Aβ42-RF) or p1541 (pRS313,CEN, URA3, CUP1::AβF19,20T/I31P-RF). While p1364 carries the wild-type Aβ42 peptide fused with the M (middle) and RF (release factor) domains of Sup35 (i.e. lacking the N-terminal prion domain), called here Aβ42-RF, p1541 carries a F19T, F20T and I31P triple mutant Aβ42 peptide that is aggregation-deficient [29], called here Aβ42m2-RF.
–
To test the effects of drugs on HDEL-Aβ42 associated toxicity, we transformed an ERG6 deleted strain, L3340, (MATα ade1-14 ura3-52 leu2-3, 112 his3-200 erg6∆::TRP1) with a 2µ gateway expression vector (pAG425 base, LEU2) carrying Aβ42 fused to the C-terminus of an ER retention signal (HDEL) expressed with the inducible GAL1 promoter, called here HDEL-Aβ42. The plasmid was generated by transferring HDEL-Aβ42 from the pAG305 GAL1::HDEL-Aβ42 integrative vector [24] to pAG425GAL-ccdB (Addgene) using Gateway Technology [63].
–
The plasmids of 12 genes previously shown to suppress HDEL-Aβ42 toxicity [24] were kindly supplied by Susan Lindquist (Whitehead Institute for Biomedical Research, Massachusetts Institute of Technology, Cambridge, MA). Each plasmid carries the yeast ORF under the control of the GAL1 promoter on the single copy plasmid, pBY011 (CEN, URA3, AmpR) [24][63].
–
Media, cultivation and transformation procedures were standard [64]. Expression of Aβ42-RF and HDEL-Aβ42 were respectively driven by the copper-inducible CUP1 promoter with 50 μM CuSO4 and the galactose inducible GAL1 promoter with 2% galactose.
–
Growth assays for the primary screen and confirmation
The primary screen employed the Prestwick Chemical Library® of 1,200 FDA approved or drug-like small compounds (Prestwick Chemical). Assays for selection of compounds that reduce the level of Aβ42-RF oligomers were as described previously [28]. Drugs in DMSO were diluted to 20 µM in assay media (2% dextrose without or with adenine + 50 µM CuSO4, respectively, for the -Ade or +Ade assay) inoculated with 1 x 105 or 1 x 104 cells/well, respectively, for the -Ade or +Ade assays. Each 384-well plate contained 32 positive (Aβ42m2-RF) and 32 negative (Aβ42-RF) controls with 0.2 μl of DMSO. The OD600 was measured after 5 days for -Ade and 4 days for +Ade plates incubated at room temperature with shaking (900 rpm). Each assay was performed in duplicate in clear flat-bottom 384-well plates (ScreenMates) using a Tecan Freedom EVO 200 liquid handling robot at the Research Resources Center at the University of Illinois, Chicago. Drug candidates that caused less than 50% and more than 70% growth in -Ade and +Ade, respectively, compared to growth in DMSO controls were selected for further analysis. Drug effects on Aβ42-RF oligomer formation were directly tested with SDS gel electrophoresis using 10% Mini-PROTEAN® TGX™ precast gels (Bio-Rad) and immunoblot analysis as described previously [65]. To detect the amount of SDS-resistant small oligomers, lysates were treated with 1% SDS for 7 min at room temperature, subjected to immunoblot analysis, and probed with antibodies against Sup35’s RF domain (BE4, developed by Viravan Prapapanich, a former post doc in our laboratory).
–
Aβ42 in vitro oligomerization and fibrilization
Aβ42 was polymerized using conditions favorable for the formation of Aβ42 small soluble oligomers [38] or fibrils [42]. For Aβ42 soluble oligomer formation, synthetic recombinant peptide (HFIP, Rpeptide) dissolved to 5 mM in anhydrous DMSO (Sigma) and sonicated was diluted to 200 µM in PBS (20 mM NaH2PO4, 140 mM NaCl, pH 7.4) supplemented with 0.2% SDS and allowed to oligomerize for 24 hrs at 37°C without agitation, in the absence (DMSO control), or presence of 100 µM drug. Finally, any high molecular Aβ42 aggregates formed were removed by a brief centrifugation at 2,000 rpm. For Aβ42 fibril formation, 50 µM of each drug was added to 20 µM Aβ42 peptide in Tris-buffered saline (25 mM Tris, 150 mM NaCl, pH 7.5) and incubated under Aβ42 fibrilization conditions [42]. Following incubation at 37°C for different lengths of time, 100 µl of Aβ42 was aliquoted into a 96 well plate with 200 µl of Thioflavine T (ThT) solution (7 µl ThT, 50 µM glycine, pH 7.1). Quantification of fibril formation was measured at 483 nm with excitation at 450 nm on a Spectra Max M5 plate reader (Molecular Devices).
–
PC12 cell toxicity
Treatment of rat PC12 neuronal cells with Aβ42 aggregates formed under the oligomerization condition described above in the presence of each drug and subsequent assays for cell viability were as described [41]. Briefly, PC12 cells, derived from rat adrenal medulla pheochromocytoma (American Type Culture Collection), were cultured in RPMI 1640 complete growth medium (ATCC) supplemented with horse serum and fetal bovine serum and grown to confluency at 37°C with 5% CO2. Cells were plated in tissue culture-treated flat bottom 96-well plates (Corning) at 10,000 cells/well and allowed to attach overnight before adding Aβ42 aggregates. Following incubation of cells for 24 hrs, the MTT cell viability assay was used to determine cell toxicity (Roche Applied Science). Briefly, 10 µl of MTT was added to each well and following incubation for 4 hrs at 37°C, 100 µl of solubilization solution was added and incubated overnight at 37°C. Absorbance was then recorded at 570 nm.
–
Oligomerization of TMR-labeled Aβ42
As described previously [43] a stock solution of TMR-K-Aβ42 (42 µM) in 4 M GdnCl was diluted to a final concentration of 1 µM in PBS (pH 7.4) supplemented with 1 mM EDTA and 5 mM β-mercaptoethanol. Drugs (10 µM) were added to the reaction. The fluorescence of TMR-K-Aβ42 was recorded as a function of time immediately after dilution of the TMR-Aβ42 in an Alphascan fluorometer (Photon Technology International), with excitation and emission monochromators set to 520 and 600 nm, respectively.
CONTRIBUTION
S.-K.P. and S.W.L. designed the overall experimental approach and wrote the paper. S.-K.P. performed most of the experiments. K.R. provided the compound libraries and prepared the robot liquid handling system for the HTS screen, and analyzed and placed the hits in chemical family groups. M.B. and M.V. helped with culturing and maintaining PC12 cells. All authors edited the manuscript.
References
- M.P. Lambert, A.K. Barlow, B.A. Chromy, C. Edwards, R. Freed, M. Liosatos, T.E. Morgan, I. Rozovsky, B. Trommer, K.L. Viola, P. Wals, C. Zhang, C.E. Finch, G.A. Krafft, and W.L. Klein, "Diffusible, nonfibrillar ligands derived from Aβ 1–42 are potent central nervous system neurotoxins", Proceedings of the National Academy of Sciences, vol. 95, pp. 6448-6453, 1998. http://dx.doi.org/10.1073/pnas.95.11.6448
- C.A. McLean, R.A. Cherny, F.W. Fraser, S.J. Fuller, M.J. Smith, K. Beyreuther, A.I. Bush, and C.L. Masters, "Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease.", Annals of neurology, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10589538
- C.G. Glabe, and R. Kayed, "Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis", Neurology, vol. 66, 2006. http://dx.doi.org/10.1212/01.wnl.0000192103.24796.42
- K.N. Dahlgren, A.M. Manelli, W.B. Stine, L.K. Baker, G.A. Krafft, and M.J. LaDu, "Oligomeric and Fibrillar Species of Amyloid-β Peptides Differentially Affect Neuronal Viability", Journal of Biological Chemistry, vol. 277, pp. 32046-32053, 2002. http://dx.doi.org/10.1074/jbc.M201750200
- T. Yang, S. Hong, T. O'Malley, R.A. Sperling, D.M. Walsh, and D.J. Selkoe, "New ELISAs with high specificity for soluble oligomers of amyloid β‐protein detect natural Aβ oligomers in human brain but not CSF", Alzheimer's & Dementia, vol. 9, pp. 99-112, 2013. http://dx.doi.org/10.1016/j.jalz.2012.11.005
- K.E.S. Matlack, D.F. Tardiff, P. Narayan, S. Hamamichi, K.A. Caldwell, G.A. Caldwell, and S. Lindquist, "Clioquinol promotes the degradation of metal-dependent amyloid-β (Aβ) oligomers to restore endocytosis and ameliorate Aβ toxicity", Proceedings of the National Academy of Sciences, vol. 111, pp. 4013-4018, 2014. http://dx.doi.org/10.1073/pnas.1402228111
- K.A. Bruggink, W. Jongbloed, E.A. Biemans, R. Veerhuis, J.A. Claassen, H.B. Kuiperij, and M.M. Verbeek, "Amyloid-β oligomer detection by ELISA in cerebrospinal fluid and brain tissue", Analytical Biochemistry, vol. 433, pp. 112-120, 2013. http://dx.doi.org/10.1016/j.ab.2012.09.014
- M.J. Savage, J. Kalinina, A. Wolfe, K. Tugusheva, R. Korn, T. Cash-Mason, J.W. Maxwell, N.G. Hatcher, S.J. Haugabook, G. Wu, B.J. Howell, J.J. Renger, P.J. Shughrue, and A. McCampbell, "A Sensitive Aβ Oligomer Assay Discriminates Alzheimer's and Aged Control Cerebrospinal Fluid", The Journal of Neuroscience, vol. 34, pp. 2884-2897, 2014. http://dx.doi.org/10.1523/JNEUROSCI.1675-13.2014
- K. Ono, M.M. Condron, and D.B. Teplow, "Structure–neurotoxicity relationships of amyloid β-protein oligomers", Proceedings of the National Academy of Sciences, vol. 106, pp. 14745-14750, 2009. http://dx.doi.org/10.1073/pnas.0905127106
- S. Lesné, M.T. Koh, L. Kotilinek, R. Kayed, C.G. Glabe, A. Yang, M. Gallagher, and K.H. Ashe, "RETRACTED ARTICLE: A specific amyloid-β protein assembly in the brain impairs memory", Nature, vol. 440, pp. 352-357, 2006. http://dx.doi.org/10.1038/nature04533
- M. Townsend, J.P. Cleary, T. Mehta, J. Hofmeister, S. Lesne, E. O'Hare, D.M. Walsh, and D.J. Selkoe, "Orally available compound prevents deficits in memory caused by the Alzheimer amyloid‐β oligomers", Annals of Neurology, vol. 60, pp. 668-676, 2006. http://dx.doi.org/10.1002/ana.21051
- D.M. Walsh, I. Klyubin, J.V. Fadeeva, W.K. Cullen, R. Anwyl, M.S. Wolfe, M.J. Rowan, and D.J. Selkoe, "Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo", Nature, vol. 416, pp. 535-539, 2002. http://dx.doi.org/10.1038/416535a
- A. Mudher, and S. Lovestone, "Alzheimer's disease – do tauists and baptists finally shake hands?", Trends in Neurosciences, vol. 25, pp. 22-26, 2002. http://dx.doi.org/10.1016/S0166-2236(00)02031-2
- I.F. Tsigelny, L. Crews, P. Desplats, G.M. Shaked, Y. Sharikov, H. Mizuno, B. Spencer, E. Rockenstein, M. Trejo, O. Platoshyn, J.X. Yuan, and E. Masliah, "Mechanisms of Hybrid Oligomer Formation in the Pathogenesis of Combined Alzheimer's and Parkinson's Diseases", PLoS ONE, vol. 3, pp. e3135, 2008. http://dx.doi.org/10.1371/journal.pone.0003135
- J. Laurén, D.A. Gimbel, H.B. Nygaard, J.W. Gilbert, and S.M. Strittmatter, "Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers", Nature, vol. 457, pp. 1128-1132, 2009. http://dx.doi.org/10.1038/nature07761
- A.D. Gitler, "Beer and Bread to Brains and Beyond: Can Yeast Cells Teach Us about Neurodegenerative Disease?", Neurosignals, vol. 16, pp. 52-62, 2007. http://dx.doi.org/10.1159/000109759
- D.F. Tardiff, V. Khurana, C.Y. Chung, and S. Lindquist, "From yeast to patient neurons and back again: Powerful new discovery platforms", Movement Disorders, vol. 29, pp. 1231-1240, 2014. http://dx.doi.org/10.1002/mds.25989
- B. Panaretou, and G.W. Jones, "Yeast models for amyloid disease", Essays in Biochemistry, vol. 56, pp. 85-97, 2014. http://dx.doi.org/10.1042/bse0560085
- D. Tribouillard, S. Bach, F. Gug, N. Desban, V. Beringue, T. Andrieu, D. Dormont, H. Galons, H. Laude, D. Vilette, and M. Blondel, "Using budding yeast to screen for anti-prion drugs", Biotechnology Journal, vol. 1, pp. 58-67, 2006. http://dx.doi.org/10.1002/biot.200500001
- P. Bharadwaj, L. Waddington, J. Varghese, and I.G. Macreadie, "A new method to measure cellular toxicity of non-fibrillar and fibrillar Alzheimer's Abeta using yeast.", Journal of Alzheimer's disease : JAD, 2008. http://www.ncbi.nlm.nih.gov/pubmed/18376056
- J. Caine, S. Sankovich, H. Antony, L. Waddington, P. Macreadie, J. Varghese, and I. Macreadie, "Alzheimer's Aβ fused to green fluorescent protein induces growth stress and a heat shock response", FEMS Yeast Research, vol. 7, pp. 1230-1236, 2007. http://dx.doi.org/10.1111/j.1567-1364.2007.00285.x
- T. von der Haar, L. Jossé, P. Wright, J. Zenthon, and M.F. Tuite, "Development of a Novel Yeast Cell-Based System for Studying the Aggregation of Alzheimer’s Disease-Associated Aβ Peptides in vivo", Neurodegenerative Diseases, vol. 4, pp. 136-147, 2007. http://dx.doi.org/10.1159/000101838
- F. D'Angelo, H. Vignaud, J. Di Martino, B. Salin, A. Devin, C. Cullin, and C. Marchal, "A yeast model for amyloid-β aggregation exemplifies the role of membrane trafficking and PICALM in cytotoxicity", Disease Models & Mechanisms, 2012. http://dx.doi.org/10.1242/dmm.010108
- S. Treusch, S. Hamamichi, J.L. Goodman, K.E.S. Matlack, C.Y. Chung, V. Baru, J.M. Shulman, A. Parrado, B.J. Bevis, J.S. Valastyan, H. Han, M. Lindhagen-Persson, E.M. Reiman, D.A. Evans, D.A. Bennett, A. Olofsson, P.L. DeJager, R.E. Tanzi, K.A. Caldwell, G.A. Caldwell, and S. Lindquist, "Functional Links Between Aβ Toxicity, Endocytic Trafficking, and Alzheimer’s Disease Risk Factors in Yeast", Science, vol. 334, pp. 1241-1245, 2011. http://dx.doi.org/10.1126/science.1213210
- S. Seshadri, "Genome-wide Analysis of Genetic Loci Associated With Alzheimer Disease", JAMA, vol. 303, pp. 1832, 2010. http://dx.doi.org/10.1001/jama.2010.574
- J. Lambert, G. Ivosev, A.L. Couzens, B. Larsen, M. Taipale, Z. Lin, Q. Zhong, S. Lindquist, M. Vidal, R. Aebersold, T. Pawson, R. Bonner, S. Tate, and A. Gingras, "Mapping differential interactomes by affinity purification coupled with data-independent mass spectrometry acquisition", Nature Methods, vol. 10, pp. 1239-1245, 2013. http://dx.doi.org/10.1038/nmeth.2702
- I. Macreadie, M. Lotfi-Miri, S. Mohotti, D. Shapira, L. Bennett, and J. Varghese, "Validation of folate in a convenient yeast assay suited for identification of inhibitors of Alzheimer's amyloid-beta aggregation.", Journal of Alzheimer's disease : JAD, 2008. http://www.ncbi.nlm.nih.gov/pubmed/18997292
- S. Park, S.D. Pegan, A.D. Mesecar, L.M. Jungbauer, M.J. LaDu, and S.W. Liebman, "Development and validation of a yeast high-throughput screen for inhibitors of Aβ42 oligomerization", Disease Models & Mechanisms, vol. 4, pp. 822-831, 2011. http://dx.doi.org/10.1242/dmm.007963
- S. Bagriantsev, and S. Liebman, "Modulation of Aβ42low-n oligomerization using a novel yeast reporter system", BMC Biology, vol. 4, 2006. http://dx.doi.org/10.1186/1741-7007-4-32
- O. Wright, L. Zhang, Y. Liu, T. Yoshimi, Y. Zheng, and A. Tunnacliffe, "Critique of the use of fluorescence‐based reporters in Escherichia coli as a screening tool for the identification of peptide inhibitors of Aβ42 aggregation", Journal of Peptide Science, vol. 19, pp. 74-83, 2012. http://dx.doi.org/10.1002/psc.2474
- T. Hamaguchi, K. Ono, A. Murase, and M. Yamada, "Phenolic Compounds Prevent Alzheimer’s Pathology through Different Effects on the Amyloid-β Aggregation Pathway", The American Journal of Pathology, vol. 175, pp. 2557-2565, 2009. http://dx.doi.org/10.2353/ajpath.2009.090417
- M. Necula, R. Kayed, S. Milton, and C.G. Glabe, "Small Molecule Inhibitors of Aggregation Indicate That Amyloid β Oligomerization and Fibrillization Pathways Are Independent and Distinct", Journal of Biological Chemistry, vol. 282, pp. 10311-10324, 2007. http://dx.doi.org/10.1074/jbc.M608207200
- M. Necula, L. Breydo, S. Milton, R. Kayed, W.E. van der Veer, P. Tone, and C.G. Glabe, "Methylene Blue Inhibits Amyloid Aβ Oligomerization by Promoting Fibrillization", Biochemistry, vol. 46, pp. 8850-8860, 2007. http://dx.doi.org/10.1021/bi700411k
- J. Chen, A.H. Armstrong, A.N. Koehler, and M.H. Hecht, "Small Molecule Microarrays Enable the Discovery of Compounds That Bind the Alzheimer’s Aβ Peptide and Reduce its Cytotoxicity", Journal of the American Chemical Society, vol. 132, pp. 17015-17022, 2010. http://dx.doi.org/10.1021/ja107552s
- E.B. Lee, L.Z. Leng, B. Zhang, L. Kwong, J.Q. Trojanowski, T. Abel, and V.M. Lee, "Targeting Amyloid-β Peptide (Aβ) Oligomers by Passive Immunization with a Conformation-selective Monoclonal Antibody Improves Learning and Memory in Aβ Precursor Protein (APP) Transgenic Mice", Journal of Biological Chemistry, vol. 281, pp. 4292-4299, 2006. http://dx.doi.org/10.1074/jbc.M511018200
- J. Zhang, T.D. Chung, and K.R. Oldenburg, "A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays", SLAS Discovery, vol. 4, pp. 67-73, 1999. http://dx.doi.org/10.1177/108705719900400206
- M.E. Bradley, S. Bagriantsev, N. Vishveshwara, and S.W. Liebman, "Guanidine reduces stop codon read‐through caused by missense mutations in SUP35 or SUP45", Yeast, vol. 20, pp. 625-632, 2003. http://dx.doi.org/10.1002/yea.985
- S. Barghorn, V. Nimmrich, A. Striebinger, C. Krantz, P. Keller, B. Janson, M. Bahr, M. Schmidt, R.S. Bitner, J. Harlan, E. Barlow, U. Ebert, and H. Hillen, "Globular amyloid β‐peptide1−42 oligomer − a homogenous and stable neuropathological protein in Alzheimer's disease", Journal of Neurochemistry, vol. 95, pp. 834-847, 2005. http://dx.doi.org/10.1111/j.1471-4159.2005.03407.x
- M.P. Lambert, K.L. Viola, B.A. Chromy, L. Chang, T.E. Morgan, J. Yu, D.L. Venton, G.A. Krafft, C.E. Finch, and W.L. Klein, "Vaccination with soluble Aβ oligomers generates toxicity‐neutralizing antibodies", Journal of Neurochemistry, vol. 79, pp. 595-605, 2001. http://dx.doi.org/10.1046/j.1471-4159.2001.00592.x
- J.P. Cleary, D.M. Walsh, J.J. Hofmeister, G.M. Shankar, M.A. Kuskowski, D.J. Selkoe, and K.H. Ashe, "Natural oligomers of the amyloid-β protein specifically disrupt cognitive function", Nature Neuroscience, vol. 8, pp. 79-84, 2004. http://dx.doi.org/10.1038/nn1372
- A.F. McKoy, J. Chen, T. Schupbach, and M.H. Hecht, "A Novel Inhibitor of Amyloid β (Aβ) Peptide Aggregation", Journal of Biological Chemistry, vol. 287, pp. 38992-39000, 2012. http://dx.doi.org/10.1074/jbc.M112.348037
- W.B. Stine, K.N. Dahlgren, G.A. Krafft, and M.J. LaDu, "In Vitro Characterization of Conditions for Amyloid-β Peptide Oligomerization and Fibrillogenesis", Journal of Biological Chemistry, vol. 278, pp. 11612-11622, 2003. http://dx.doi.org/10.1074/jbc.M210207200
- K. Garai, and C. Frieden, "Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ", Proceedings of the National Academy of Sciences, vol. 110, pp. 3321-3326, 2013. http://dx.doi.org/10.1073/pnas.1222478110
- I. Benilova, E. Karran, and B. De Strooper, "The toxic Aβ oligomer and Alzheimer's disease: an emperor in need of clothes", Nature Neuroscience, vol. 15, pp. 349-357, 2012. http://dx.doi.org/10.1038/nn.3028
- P.N. Lacor, M.C. Buniel, P.W. Furlow, A. Sanz Clemente, P.T. Velasco, M. Wood, K.L. Viola, and W.L. Klein, "Aβ Oligomer-Induced Aberrations in Synapse Composition, Shape, and Density Provide a Molecular Basis for Loss of Connectivity in Alzheimer's Disease", The Journal of Neuroscience, vol. 27, pp. 796-807, 2007. http://dx.doi.org/10.1523/JNEUROSCI.3501-06.2007
- J. Lambert, . , C.A. Ibrahim-Verbaas, D. Harold, A.C. Naj, R. Sims, C. Bellenguez, G. Jun, A.L. DeStefano, J.C. Bis, G.W. Beecham, B. Grenier-Boley, G. Russo, T.A. Thornton-Wells, N. Jones, A.V. Smith, V. Chouraki, C. Thomas, M.A. Ikram, D. Zelenika, B.N. Vardarajan, Y. Kamatani, C. Lin, A. Gerrish, H. Schmidt, B. Kunkle, M.L. Dunstan, A. Ruiz, M. Bihoreau, S. Choi, C. Reitz, F. Pasquier, P. Hollingworth, A. Ramirez, O. Hanon, A.L. Fitzpatrick, J.D. Buxbaum, D. Campion, P.K. Crane, C. Baldwin, T. Becker, V. Gudnason, C. Cruchaga, D. Craig, N. Amin, C. Berr, O.L. Lopez, P.L. De Jager, V. Deramecourt, J.A. Johnston, D. Evans, S. Lovestone, L. Letenneur, F.J. Morón, D.C. Rubinsztein, G. Eiriksdottir, K. Sleegers, A.M. Goate, N. Fiévet, M.J. Huentelman, M. Gill, K. Brown, M.I. Kamboh, L. Keller, P. Barberger-Gateau, B. McGuinness, E.B. Larson, R. Green, A.J. Myers, C. Dufouil, S. Todd, D. Wallon, S. Love, E. Rogaeva, J. Gallacher, P. St George-Hyslop, J. Clarimon, A. Lleo, A. Bayer, D.W. Tsuang, L. Yu, M. Tsolaki, P. Bossù, G. Spalletta, P. Proitsi, J. Collinge, S. Sorbi, F. Sanchez-Garcia, N.C. Fox, J. Hardy, M.C.D. Naranjo, P. Bosco, R. Clarke, C. Brayne, D. Galimberti, M. Mancuso, F. Matthews, S. Moebus, P. Mecocci, M. Del Zompo, W. Maier, H. Hampel, A. Pilotto, M. Bullido, F. Panza, P. Caffarra, B. Nacmias, J.R. Gilbert, M. Mayhaus, L. Lannfelt, H. Hakonarson, S. Pichler, M.M. Carrasquillo, M. Ingelsson, D. Beekly, V. Alvarez, F. Zou, O. Valladares, S.G. Younkin, E. Coto, K.L. Hamilton-Nelson, W. Gu, C. Razquin, P. Pastor, I. Mateo, M.J. Owen, K.M. Faber, P.V. Jonsson, O. Combarros, M.C. O'Donovan, L.B. Cantwell, H. Soininen, D. Blacker, S. Mead, T.H. Mosley, D.A. Bennett, T.B. Harris, L. Fratiglioni, C. Holmes, R.F.A.G. de Bruijn, P. Passmore, T.J. Montine, K. Bettens, J.I. Rotter, A. Brice, K. Morgan, T.M. Foroud, W.A. Kukull, D. Hannequin, J.F. Powell, M.A. Nalls, K. Ritchie, K.L. Lunetta, J.S.K. Kauwe, E. Boerwinkle, M. Riemenschneider, M. Boada, M. Hiltunen, E.R. Martin, R. Schmidt, D. Rujescu, L. Wang, J. Dartigues, R. Mayeux, C. Tzourio, A. Hofman, M.M. Nöthen, C. Graff, B.M. Psaty, L. Jones, J.L. Haines, P.A. Holmans, M. Lathrop, M.A. Pericak-Vance, L.J. Launer, L.A. Farrer, C.M. van Duijn, C. Van Broeckhoven, V. Moskvina, S. Seshadri, J. Williams, G.D. Schellenberg, P. Amouyel, . , . , and . , "Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer's disease", Nature Genetics, vol. 45, pp. 1452-1458, 2013. http://dx.doi.org/10.1038/ng.2802
- . Ganjei, J.K., "Targeting amyloid precursor protein secretases: Alzheimer's disease and beyond", Drug News & Perspectives, vol. 23, pp. 573, 2010. http://dx.doi.org/10.1358/dnp.2010.23.9.1507297
- H. Zheng, and E.H. Koo, "Array", Molecular Neurodegeneration, vol. 1, pp. 5, 2006. http://dx.doi.org/10.1186/1750-1326-1-5
- Y. Wang, H. Yin, L. Wang, A. Shuboy, J. Lou, B. Han, X. Zhang, and J. Li, "Curcumin as a Potential Treatment for Alzheimer's Disease: A Study of the Effects of Curcumin on Hippocampal Expression of Glial Fibrillary Acidic Protein", The American Journal of Chinese Medicine, vol. 41, pp. 59-70, 2013. http://dx.doi.org/10.1142/S0192415X13500055
- F. Yang, G.P. Lim, A.N. Begum, O.J. Ubeda, M.R. Simmons, S.S. Ambegaokar, P.P. Chen, R. Kayed, C.G. Glabe, S.A. Frautschy, and G.M. Cole, "Curcumin Inhibits Formation of Amyloid β Oligomers and Fibrils, Binds Plaques, and Reduces Amyloid in Vivo", Journal of Biological Chemistry, vol. 280, pp. 5892-5901, 2005. http://dx.doi.org/10.1074/jbc.M404751200
- Y. Feng, X. Wang, S. Yang, Y. Wang, X. Zhang, X. Du, X. Sun, M. Zhao, L. Huang, and R. Liu, "Resveratrol inhibits beta-amyloid oligomeric cytotoxicity but does not prevent oligomer formation", NeuroToxicology, vol. 30, pp. 986-995, 2009. http://dx.doi.org/10.1016/j.neuro.2009.08.013
- D.E. Ehrnhoefer, J. Bieschke, A. Boeddrich, M. Herbst, L. Masino, R. Lurz, S. Engemann, A. Pastore, and E.E. Wanker, "EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers", Nature Structural & Molecular Biology, vol. 15, pp. 558-566, 2008. http://dx.doi.org/10.1038/nsmb.1437
- Q. Nie, X. Du, and M. Geng, "Small molecule inhibitors of amyloid β peptide aggregation as a potential therapeutic strategy for Alzheimer's disease", Acta Pharmacologica Sinica, vol. 32, pp. 545-551, 2011. http://dx.doi.org/10.1038/aps.2011.14
- R.A. Cherny, C.S. Atwood, M.E. Xilinas, D.N. Gray, W.D. Jones, C.A. McLean, K.J. Barnham, I. Volitakis, F.W. Fraser, Y. Kim, X. Huang, L.E. Goldstein, R.D. Moir, J.T. Lim, K. Beyreuther, H. Zheng, R.E. Tanzi, C.L. Masters, and A.I. Bush, "Treatment with a Copper-Zinc Chelator Markedly and Rapidly Inhibits β-Amyloid Accumulation in Alzheimer's Disease Transgenic Mice", Neuron, vol. 30, pp. 665-676, 2001. http://dx.doi.org/10.1016/S0896-6273(01)00317-8
- C.W. Ritchie, A.I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. MacGregor, L. Kiers, R. Cherny, Q. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R.E. Tanzi, and C.L. Masters, "Metal-Protein Attenuation With Iodochlorhydroxyquin (Clioquinol) Targeting Aβ Amyloid Deposition and Toxicity in Alzheimer Disease", Archives of Neurology, vol. 60, pp. 1685, 2003. http://dx.doi.org/10.1001/archneur.60.12.1685
- E.L. Sampson, L. Jenagaratnam, and R. McShane, "Metal protein attenuating compounds for the treatment of Alzheimer's dementia", Cochrane Database of Systematic Reviews, vol. 2014, 2014. http://dx.doi.org/10.1002/14651858.CD005380.pub5
- K. Ma, L.A. Thomason, and J. McLaurin, "scyllo-Inositol, Preclinical, and Clinical Data for Alzheimer’s Disease", Current State of Alzheimer's Disease Research and Therapeutics, pp. 177-212, 2012. http://dx.doi.org/10.1016/B978-0-12-394816-8.00006-4
- C.J. Garwood, J.D. Cooper, D.P. Hanger, and W. Noble, "Anti-Inflammatory Impact of Minocycline in a Mouse Model of Tauopathy", Frontiers in Psychiatry, vol. 1, 2010. http://dx.doi.org/10.3389/fpsyt.2010.00136
- C.J. Garwood, A.M. Pooler, J. Atherton, D.P. Hanger, and W. Noble, "Astrocytes are important mediators of Aβ-induced neurotoxicity and tau phosphorylation in primary culture", Cell Death & Disease, vol. 2, pp. e167-e167, 2011. http://dx.doi.org/10.1038/cddis.2011.50
- W. Noble, C. Garwood, J. Stephenson, A.M. Kinsey, D.P. Hanger, and B.H. Anderton, "Minocycline reduces the development of abnormal tau species in models of Alzheimer's disease", The FASEB Journal, vol. 23, pp. 739-750, 2008. http://dx.doi.org/10.1096/fj.08-113795
- W. Noble, C.J. Garwood, and D.P. Hanger, "Minocycline as a potential therapeutic agent in neurodegenerative disorders characterized by protein misfolding", Prion, vol. 3, pp. 78-83, 2009. http://dx.doi.org/10.4161/pri.3.2.8820
- M.J. Guerrero-Muñoz, D.L. Castillo-Carranza, S. Krishnamurthy, A.A. Paulucci-Holthauzen, U. Sengupta, C.A. Lasagna-Reeves, Y. Ahmad, G.R. Jackson, and R. Kayed, "Amyloid-β oligomers as a template for secondary amyloidosis in Alzheimer's disease", Neurobiology of Disease, vol. 71, pp. 14-23, 2014. http://dx.doi.org/10.1016/j.nbd.2014.08.008
- S. Alberti, A.D. Gitler, and S. Lindquist, "A suite of Gateway® cloning vectors for high‐throughput genetic analysis in Saccharomyces cerevisiae", Yeast, vol. 24, pp. 913-919, 2007. http://dx.doi.org/10.1002/yea.1502
- F. Sherman, "Getting started with yeast", Guide to Yeast Genetics and Molecular and Cell Biology - Part B, pp. 3-41, 2002. http://dx.doi.org/10.1016/s0076-6879(02)50954-x
- S.N. Bagriantsev, V.V. Kushnirov, and S.W. Liebman, "Analysis of Amyloid Aggregates Using Agarose Gel Electrophoresis", Methods in Enzymology, pp. 33-48, 2006. http://dx.doi.org/10.1016/S0076-6879(06)12003-0
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
This work was supported by grants to S.W.L. from the Alzheimer’s Association (IIRG-06-25468 and IIRG-10-173736) and the NIH (R21 AG02881). The authors are grateful to Dr. Susan Lindquist (MIT, Cambridge, MA), Dr. Arron Gitler (Stanford University, San Francisco, CA), and Dr. Virginia Lee (University of Pennsylvania, Philadelphia, PA) for kindly providing a yeast HDEL-Aβ42 toxic strain, plasmids carrying yeast ORFs under control of the GAL1 promoter, and NAB61 antibody respectively. We also thank Dr. Andrew Mesecar (Purdue University, Indiana, IN) and Dr. Scott Pegan (University of Georgia, Athens, GA) for their ideas and helpful suggestions. Finally, we thank Dr. Kanchan Garai and Dr. Carl Frieden (Washington University School of Medicine, St. Louis, MO) for testing and providing data of in vitro assay for oligomerization of TMR-labeled Aβ42, which is shown in Fig. 4D.
COPYRIGHT
© 2016

Inhibition of Aβ42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs by Sei-Kyoung Park et al. is licensed under a Creative Commons Attribution 4.0 International License.








