
Research Articles:
Microbial Cell, Vol. 5, No. 4, pp. 169 - 183; doi: 10.15698/mic2018.04.623
Molecular signature of the imprintosome complex at the mating-type locus in fission yeast
1 Genomes and Genetics department, Genome Dynamics Unit, UMR 3525 CNRS, Institut Pasteur, 25-28 rue du docteur Roux, Paris, France. Sorbonne Universités, Université Pierre et Marie Curie, Institut de Formation Doctorale, 75252 Paris Cedex 05, France.
2 Center for Human Immunology, CRT & Hub de Bioinformatique et Biostatistiques, C3BI, Institut Pasteur, 25-28 rue du Docteur Roux, Paris, France.
3 Genomes and Genetics department, Plate-forme Transcriptome & Epigenome, Biomics, Centre d’Innovation et Recherche Technologique (Citech), Institut Pasteur, 25-28 rue du Docteur Roux, Paris, France.
4 Enseignement, Institut Pasteur, 25-28 rue du Docteur Roux, Paris, France.
Keywords: imprint, mating type switching, epigenetics, replication, Lsd1, Lsd2, Sap1.
Abbreviations:
ChiP-seq – chromatin-immunoprecipitation and whole genome sequencing,
DSB – double-strand break,
HMG – high mobility group,
M – minus,
MT – mating type,
P – plus.
Received originally: 05/09/2017 Received in revised form: 21/12/2017
Accepted: 02/01/2018
Published: 16/01/2018
Correspondence:
Benoit Arcangioli, Genomes and Genetics department, Genome Dynamics Unit, UMR 3525 CNRS, Institut Pasteur, 25-28 rue du docteur Roux, Paris, France. Sorbonne Universités, Université Pierre et Marie Curie, Institut de Formation Doctorale, 75252 Paris Cedex 05, France; Tel: 0033145688454; benoit.arcangioli@pasteur.fr
Conflict of interest statement: The authors declare that no competing interests exist.
Please cite this article as: Célia Raimondi, Bernd Jagla, Caroline Proux, Hervé Waxin, Serge Gangloff, Benoit Arcangioli (2018). Molecular signature of the imprintosome complex at the mating-type locus in fission yeast. Microbial Cell 5(4): 169-183. doi: 10.15698/mic2018.04.623
Abstract
Genetic and molecular studies have indicated that an epigenetic imprint at mat1, the sexual locus of fission yeast, initiates mating type switching. The polar DNA replication of mat1 generates an imprint on the Watson strand. The process by which the imprint is formed and maintained through the cell cycle remains unclear. To understand better the mechanism of imprint formation and stability, we characterized the recruitment of early players of mating type switching at the mat1 region. We found that the switch activating protein 1 (Sap1) is preferentially recruited inside the mat1M allele on a sequence (SS13) that enhances the imprint. The lysine specific demethylases, Lsd1/2, that control the replication fork pause at MPS1 and the formation of the imprint are specifically drafted inside of mat1, regardless of the allele. The CENP-B homolog, Abp1, is highly enriched next to mat1 but it is not required in the process. Additionally, we established the computational signature of the imprint. Using this signature, we show that both sides of the imprinted molecule are bound by Lsd1/2 and Sap1, suggesting a nucleoprotein protective structure defined as imprintosome.
INTRODUCTION
Haploid Schizosaccharomyces pombe cells exist as two mating types (MTs), P (for plus) and M (for minus), that switch during cell divisions. The MT of the cell is determined by the allele (M or P) expressed at the mat1 locus on the right arm of chromosome II, 500 kb away from the centromere: mat1P in P cells and mat1M in M cells [1][2]. The mat1 allele can be replaced efficiently by genetic information contained in one of the two silent donor cassettes mat2P and mat3M [2] [3][4]. mat2P and mat3M are located 17 kb and 29 kb centromere-distal to mat1, respectively [3][5], and are maintained in a silent chromatin state preventing their transcription or recombination [5][6][7]. Two inverted repeats flank the donor region (IR-L and IR-R) and act as boundary elements [8][9]. Additionally, a centromeric repeated sequence located between mat2P and mat3M, CEN-H, promotes the formation of heterochromatin ([10], see Figure 1). A salient feature of the MT loci is that they are flanked by homologous sequences. The H1 homology box (59 bp) is located on the centromere distal (right) side of the cassettes and the H2 homology box (135 bp) on the centromere proximal (left) side [2]. Both sequences are thought to be essential for base pairing during the initiation and resolution steps of the gene conversion process required for MT switching [11][12]. The H3 box (57 bp) is located to the left of H2 at mat2 and mat3 [2], and is not required for MT switching [13].
–
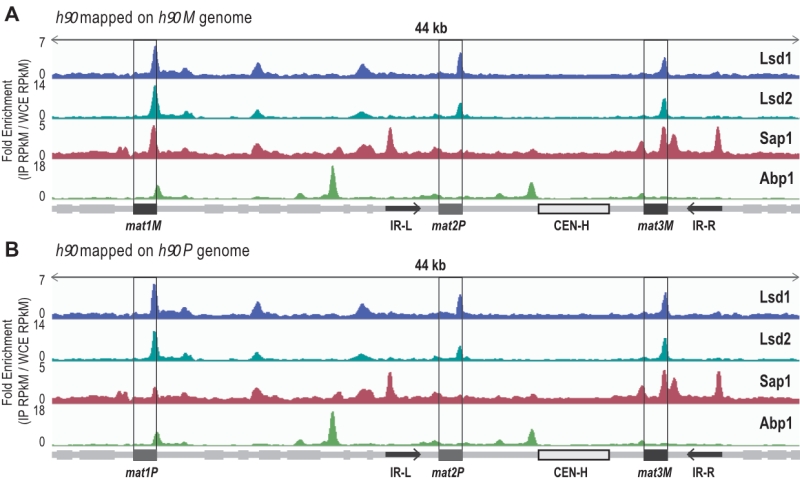 | FIGURE 1: Lsd1, Lsd2, Sap1 and Abp1 are recruited to the mating type region. (A) Distribution of normalized enrichments of the Abp1, Lsd1, Lsd2, and Sap1 ChIPs in a h90 strain (IP RPkM reads per kilobase million/ WCE RPkM). The sequence used for the alignment is a 44-kb region that contains the MT region with the M allele at mat1: h90 M. The 3 mat loci are indicated as well as the CEN-H region and the inverted repeats IR-L and IR-R. Light gray boxes show genes present in this region. (B) Same than in A) except that the sequence used for the alignment is a 44-kb region that contains the MT region with the P allele at mat1: h90 P. |
Extensive pedigree analysis at the single-cell level demonstrated that two consecutive divisions are required to produce one switched cell among four related cousins. Fission yeast possesses a remarkable genetically programmed system initiated by a site and DNA strand-specific imprint at the mat1 locus for changing its MT (reviewed in [14][15][16]). The first division produces the imprint on one of the sister chromatids at mat1, while the second division triggers a double-strand break (DSB) on the imprinted chromatid that initiates MT switching [17].
–
It was shown that the polarity of DNA replication of mat1 is instrumental in the establishment and the strand specificity of the imprint [18]. Two sites control the replication polarity at mat1, RTS1 that blocks the fork coming in from the centromere and MPS1 located near the site of the imprint that pauses the fork coming in from the right. The pausing of the replication fork at MPS1 is a prerequisite for the formation of the imprint [14]. Thus, the imprint is made on the newly synthesized lagging strand during the resumption of DNA synthesis at MPS1 [17][19].
–
Molecular studies have suggested that the imprint at mat1 is either a single-strand DNA break [20] or one or two ribonucleotides [21]. The imprint was mapped to the Watson strand corresponding to the neo-synthesized lagging strand, at the junction of the mat1 allele and the H1 homology box [22]. Furthermore, the position of the imprint is site-specific but sequence-independent [19][20]. Interestingly, the position of the break on the Watson strand at mat1 differs by three nucleotides between the mat1P and mat1M alleles [22], indicating that the position is dictated from within the mat1M and mat1P sequences. The imprint is protected against DNA repair and remains stable throughout the entire length of the cell cycle to be transiently converted to a polar DSB during the following S-phase [17][20]. Repair of this break is not random; mat1P prefers mat3M, and mat1M prefers the nearby mat2P in 90% of switches. This preference is called directionality of switching [23]. In strains in which the donor sequences are deleted [24], the position and level of the imprint are identical to that in wild type, but the DSB is repaired off the sister [24][25].
–
Several cis-acting sequences are required for the formation and maintenance of the imprint. The SS2 and SS13 deletions are located within mat1M. SS2 is important for the pause and the imprint, whereas SS13 acts as a spacer element that allows efficient imprinting [19]. To the right of mat1M, the Msmt-0 deletion maintains efficient pausing but abolishes the imprint [18][26]. The Lsd1/2 (Lysine Specific Demethylases) and the Swi1/Swi3 (Switch gene) complexes are necessary for the pause and the subsequent imprint [17][27][28]. Lsd1 is part of a complex including Lsd2 as well as Phf1/Phf2, two plant homeodomain finger proteins [29]. The complex also associates with topoisomerase 2 (Top2), replication factor A (Rfa1) and Sap1 [29], essential proteins with a role in DNA replication. The involvement of Lsd1 in DNA replication and DNA damage response is not new [30], and we showed that Lsd1/2 act redundantly to promote the pause through both their HMG (high mobility group) domain and amine oxidase activity. In addition, the recruitment of Lsd1/2 to mat1 requires SS2 [27]. Swi1/3 proteins are associated with the replication fork and stabilize it during the pause [28][31]. Sap1, the Switch Activating Protein 1 is an essential gene that interacts with SAS1, a sequence covered by the Msmt-0 deletion [32]. Sap1 was also described to play a role in DNA replication [33][34][35][36], retrotransposition targeting [37][38] and chromosomal organization [39][40]. Finally, Abp1 (ARS Binding Protein 1), a CENP-B (Centromeric Protein B) homolog that antagonizes Sap1 at LTRs (long terminal repeat), is enriched next to SAS1, to the right of mat1M [35]. Abp1 was proposed to control the directionality of switching by regulating the alternative expression of the swi2 gene (Switch gene 2) [41][42][43], important for the spreading of the Swi2/Swi5 recombination-promoting complex on the mat2/3 region [43].
–
To advance our understanding of the process of imprint formation and stability, we analyzed by chromatin-immunoprecipitation and whole genome sequencing (ChIP-seq) the recruitment of Abp1, Lsd1, Lsd2, and Sap1 proteins to the mat1 region. Because the Lsd1/2 complex interacts with a cis-acting element located within the MT cassettes and promotes replication pause at mat1 but not at mat2P and mat3M, we compared the interactions of the four proteins in the wild-type strain and in two strains containing a deletion of both the mat2P and mat3M cassettes. Thus, we have established the computational signature of the imprint and propose a model in which Lsd1 and Lsd2 are exclusively recruited to the mat1 locus.
RESULTS
ChIP-sequencing mapping of Abp1, Lsd1/2 and Sap1 to the h90 mating type region
It has been previously reported that Abp1, Lsd1/2 and Sap1 bind to the mat1 region [27][35] [44]. However, several limitations arose from those studies: The strains that were initially used to map Abp1 and Sap1 by ChIP-seq were rearranged in the MT region (h-S), and the length of the reads (30 nucleotides) prevented their unambiguous assignment to mat1, 2 or 3 cassettes [35]. Concerning Lsd1/2, whole genome analyses had been achieved by ChIP-chip [45], but no detailed analysis of the MT region was performed. More recently, we mapped Lsd1/2 to mat1 by ChIP-qPCR [27].
–
To extend the mapping of these proteins to the entire MT region, we performed ChIP-seq of Abp1, Lsd1/2 and Sap1 on the wild-type and switching proficient h90 strain (Figure 1). To achieve proper mapping, we constructed two reference genomes carrying either the M or P allele at mat1 (Figure 1). We found that both Lsd1 and Lsd2 are enriched at the mat1, 2 and 3 cassettes, whereas Sap1 was more enriched at the mat1M and mat3M cassettes than mat1P and mat2P (Figure 1). Additionally, Sap1 was found to be enriched at the IR-L and IR-R boundary elements [8][46]. Concerning Abp1, we confirmed that it is indeed enriched at the left border of CEN-H [41][47]. Interestingly, we found that Abp1 is also enriched at the right border of mat1 in an h90 background. This observation prompted us to investigate the potential involvement of Abp1 in the early steps of MT switching.
–
Abp1 is recruited to swi2 when M is present at mat1
Using 2D gel analysis, we addressed the role of Abp1 in MPS1 activity. Unlike the situation observed at LTRs, Abp1 is not required for the pausing activity of MPS1 (Figure 2A), consistent with Abp1 being dispensable for the formation and maintenance of the imprint [41]. Earlier work has shown that Mc, encoded from the mat1M cassette, and Abp1 control the expression of Swi2, a key player in the directionality process [42][43]. Moreover, the recruitment of Mc to swi2 depends on Abp1 [42]. Reciprocally, using the two stable M and P strains with deleted donor sequences, we found that the enrichment of Abp1 at swi2 is restricted to M cells, while the enrichment at the neighboring peak is unaffected by the MT of the cells (Figure 2B). The genome wide analysis showed that Abp1 M-specific interaction is unique for the swi2 gene.
–
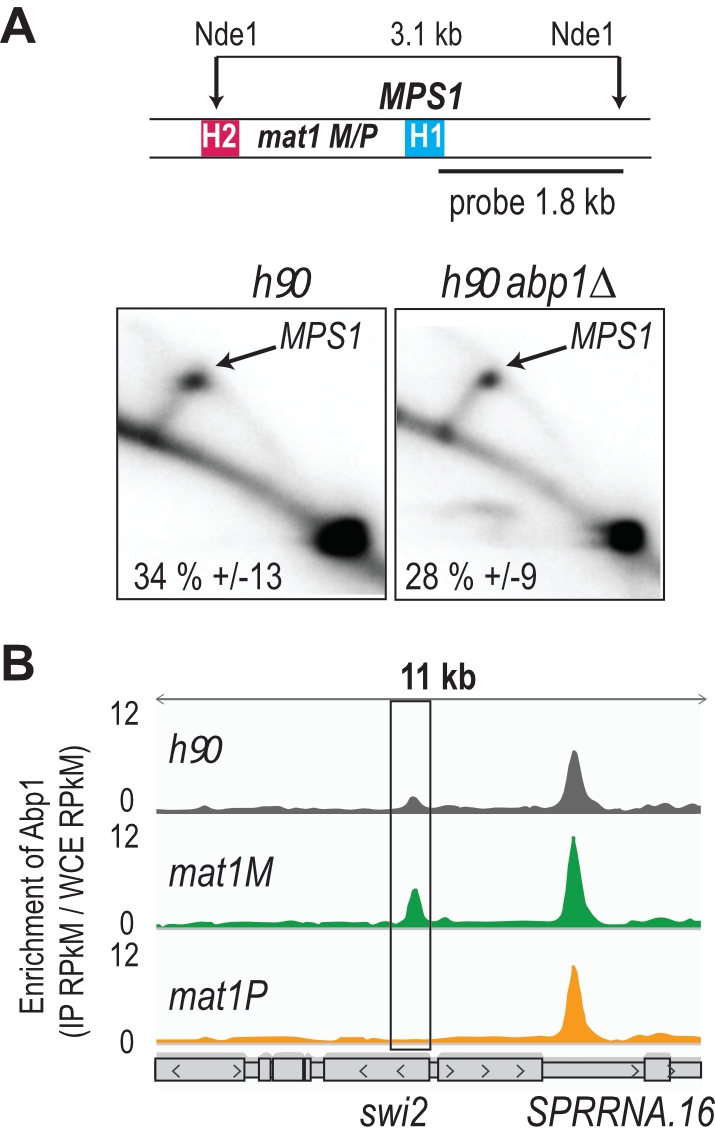 | FIGURE 2: Abp1 does not control MPS1 but is recruited at swi2 as a function of the mating type. (A) The upper panel indicates the probe and re-striction enzyme used for the analysis of the replication intermediates. The lower panel represents the 2D gel analysis of the MPS1 site at mat1 in h90 strain and in h90 abp1∆ strain (n=3). (B) Distribution of normalized enrichments of the Abp1 ChIPs at the swi2 locus (IP RPkM / WCE RPkM) in h90 (gray), mat1M stable (green) and mat1P stable (orange). Gray boxes indicate the genes in the region and arrows the transcription direction. Loci where Abp1 is enriched are annotated. |
Lsd1/2, Sap1 and Abp1 map at mat1
The enrichment positions of the four proteins and the repeated nature of the MT cassettes introduce an ambiguity that cannot be resolved during alignment. To pinpoint the Lsd1/2 and Sap1 binding sites to mat1, we used the mat1M ∆2-3 and mat1P ∆2-3 strains deleted for the donor sequences [24] (Figure 3A). Abp1 is enriched outside of the mat1 locus (to the right of the H1 cassette), and its enrichment is independent of the allele present at mat1 (Figure 3B, 3C). Lsd1 and Lsd2 are enriched both in mat1M and mat1P with Lsd2 and Sap1 more enriched in mat1M (Figure 3B, 3C).
–
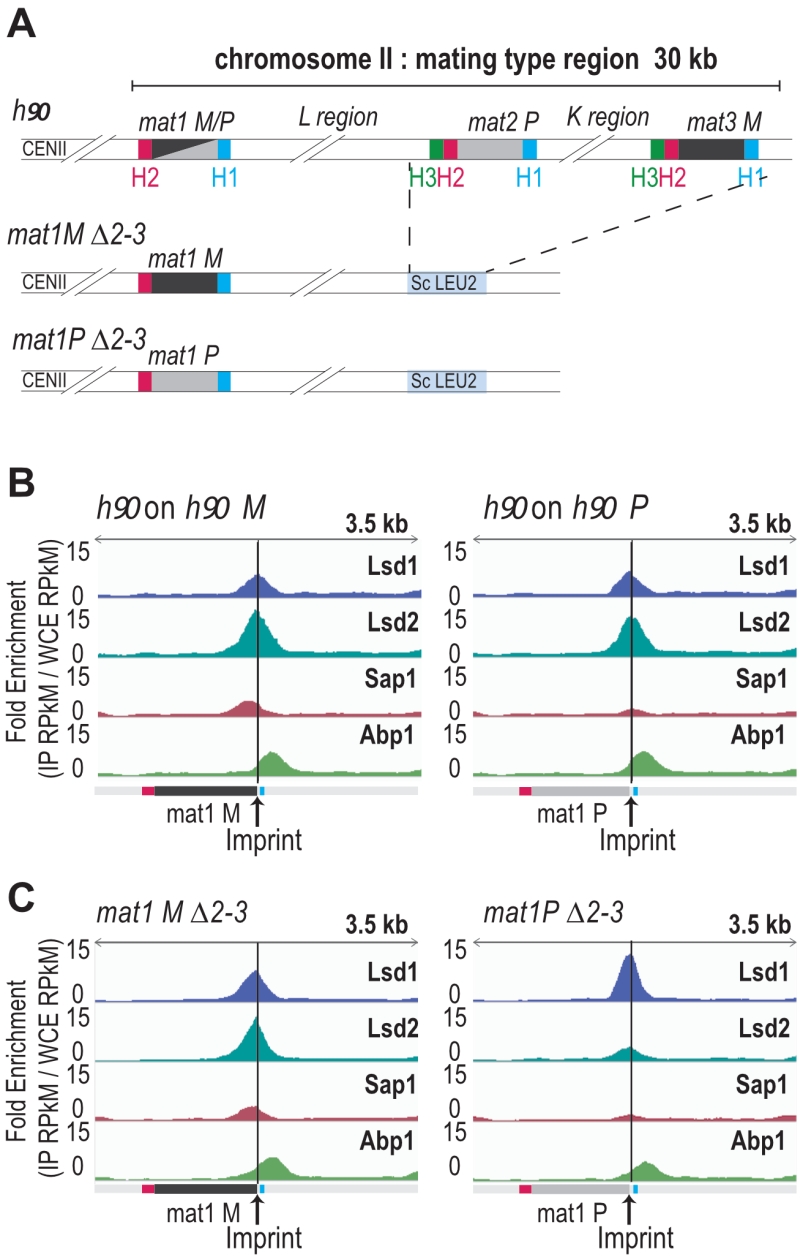 | FIGURE 3: Abp1, Lsd1/2 and Sap1 are recruited at mat1. (A) Schematic view of the strains used. The stable mat1M ∆2-3 and mat1P ∆2-3 strains as well as the wild type h90 strain are drawn. (B) On the left: distribution of normalized enrichments of the Abp1, Lsd1, Lsd2, and Sap1 ChIPs on the h90 background (IP RPkM / WCE RPkM). The sequence used for the alignment is a 44-kb region that contains the MT region with the M allele at mat1: h90 M. Only mat1M is shown. On the right: same as at the left, with the h90 P sequence used for the alignment. Only mat1P is shown. (C) Same as in (B) with the mat1M ∆2-3 background on the left and the mat1P ∆2-3 background on the right. The sequence used for the alignments is a 10-kb region containing mat1M (for the left panel) or mat1P (for the right panel).( B-C) the imprint site is indicated as well as the H2 (pink) and H1 (blue) homology boxes. |
We next analyzed the reads that can only map to a unique position on the reference genome (unique mapper) in a h90 wild type context (Figure 4A), where the donor sequences are present (Supplementary Figure 4). As expected, the highly repetitive sequences of IR-L and IR-R are no longer covered in the two h90 genomes (h90 M and h90 P) (Supplementary Figure 4A and 4B). When the M allele is at mat1, we found no unique mapper reads at mat1 and mat3 (Figure 4B). Similarly, when the P allele is at mat1, no unique mapper reads at mat1 and mat2 were found (Figure 4C). Conversely, Abp1 is located outside of the mat1 cassette in a unique region and can thus be used for alignment. In the case of Lsd1/2, the enriched unique mappers are well maintained at mat1 (both in the M and P context) and strongly diminished to nearly background level a mat2P and mat3M loci, indicating that Lsd1/2 are preferentially recruited at mat1. Furthermore, the absence of Lsd1/2 at the two silent loci was confirmed using ChIP-qPCR (Supplementary Figure 4C). The situation for Sap1 is less clear, and its profile suggests that it is recruited to M but poorly to P. This hints that Sap1 is recruited both inside mat1M and to SAS1 outside of mat1.
–
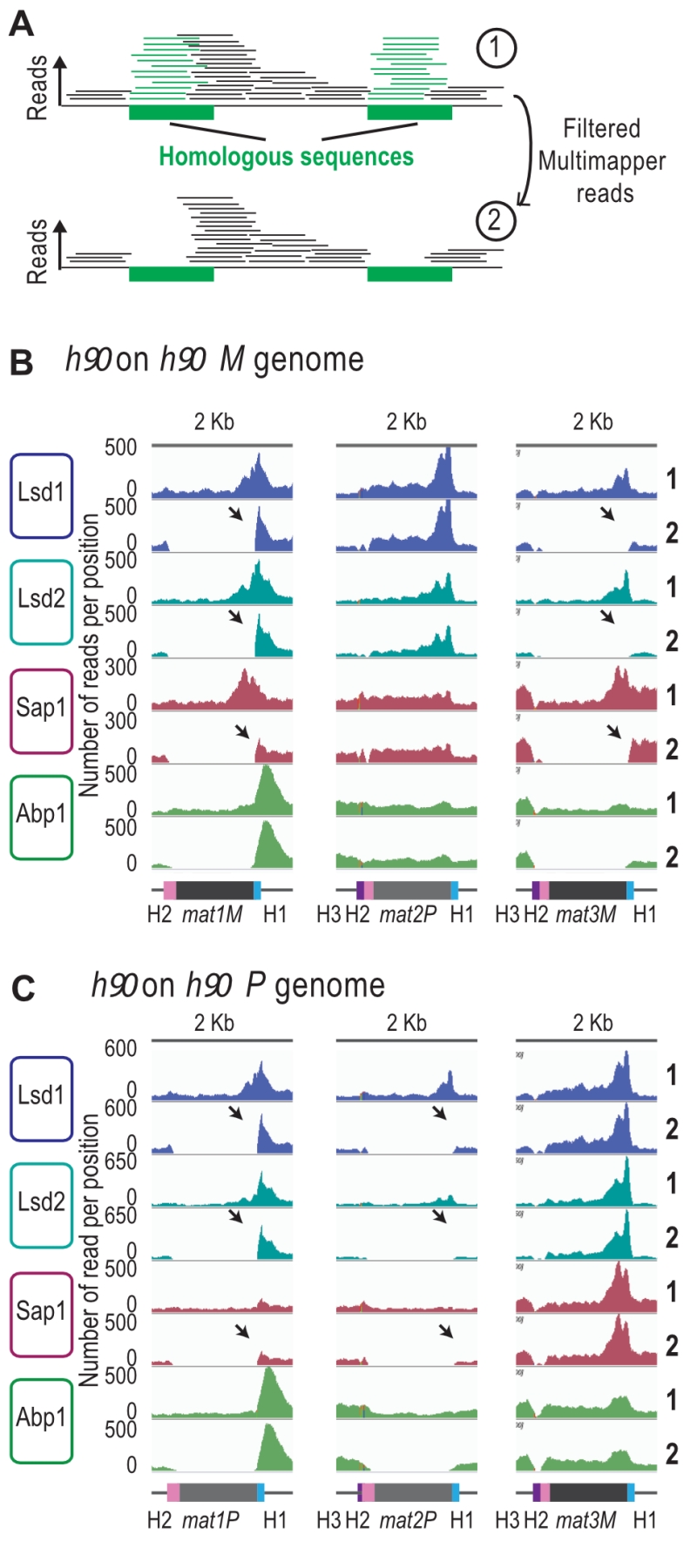 | FIGURE 4: Lsd1/2 and Abp1 are recruited only at mat1. (A) Schematic representation of the analysis used. Green boxes represent homologous sequences. Reads that can be unambiguously mapped are represented by black lines and multi-mapper reads are represented by green lines. Upper drawing represents the result of an alignment without filter (1). Lower drawing represents the alignment after that multi-mapper reads are removed using "samtools" algorithm (2). (B) Distribution of raw coverage of the Abp1, Lsd1, Lsd2, and Sap1 ChIPs. The sequence used for the alignment is a 44-kb region that contains the MT region with the M allele at mat1: h90 M. The lines numbered 1 are the coverage without filter and the lines numbered 2 are the coverage obtained after removal of multi-mapper reads. The arrows indicate the unique-mapper reads at the junction of the mat loci. (C) Same as in (B) with the h90 P sequence used for the alignment. |
Characterization of the Sap1 binding in the M allele
We used in vitro and in vivo complementary approaches to further determine Sap1 binding sequences in the M allele. The gel shift experiments were achieved using 14 double-stranded DNA probes labeled in 5′. Two positive controls were included (SAS1 and Ter1) in addition to 10 regions mapping inside the M allele and 2 mapping outside (Figure 5A). Using crude protein extracts, we observed the expected mobility shift for SAS1 and Ter1 as well as for 3 probes inside the M allele (Figure 5B). We used purified Sap1 protein to confirm that it is responsible for the shift (Figure 5C). Since M4, M5 and M6 are part of a region involved in the stability of the imprint (SS13) [19], we analyzed by ChIP-seq the enrichment of Sap1 in several donor less strains with deletions spanning the mat1M locus. The Msmt-0 deletion does not change the enrichment signal within mat1M, whereas the SS13 deletion strongly decreases the Sap1 signal, supporting the in vitro interaction result (Figure 5D Msmt-0 and SS13). The slight decrease on the right of the mat1M smt-0-containing deletion together with the remaining weak Sap1 signal in SS13 is attributed to the SAS1 interaction located 140 bp away from the imprint. The SS2 deletion abolishes DNA replication pausing and imprinting [19] but does not affect Sap1 enrichment, whose signal is slightly shifted toward the imprint (Figure 5D ∆SS2). These results indicate that Sap1 is preferentially enriched in vivo on two sequences (SS13 and SAS1) that are not necessary for replication fork pausing but required for imprint formation or maintenance, at least at the mat1M locus.
–
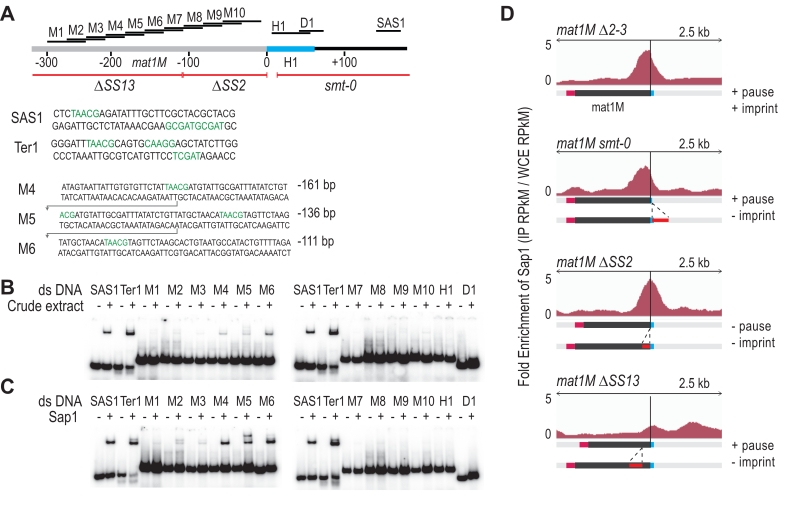 | FIGURE 5: Sap1 is recruited in mat1M. (A) Schematic view of right region of the mat1M locus. The probes used for the shift assay as well as the sites of the deletion strains used for the ChIP-seq of Sap1 are indicated. The sequence of the probes that trigger a shift in B and C are indicated. (B) Electrophoretic mobility shift analysis (EMSA) of crude extracts from S. pombe. SAS1 and Ter1 are used as controls. In lanes (-), 1 ng of radio labelled dsDNA is used with no proteins. In lanes (+), 5 µg of total proteins and 1 ng of radio labelled ds DNA are used. (C) EMSA of Sap1-6xHis protein purified from E. coli. In lanes (-), 1 ng of radio labelled ds DNA is used with no proteins. In lanes (+), 40 ng of Sap1-6xHis protein and 1 ng of radio labelled ds DNA are used. (D) Distribution of normalized enrichments of Sap1 ChIPs (IP RPkM / WCE RPkM) in the mat1M ∆2-3, mat1M smt-0 ∆2-3, mat1M ∆SS2 ∆2-3 and mat1M ∆SS13 ∆2-3 backgrounds. The sequence used for the alignment is a 10-kb region that contains the mat1M, mat1M smt-0, mat1M ∆SS2 or mat1M ∆SS13, respectively. Only mat1M is shown. The MT switching phenotype of the deletion strains is indicated. |
Detection of an imprinted site by high throughput sequencing
The imprint was described as either a single stranded break [20] or a one/two ribonucleotide(s) [21][48] that behave(s) as a mechanical fragile site [49]. Because the chromatin preparation and the Illumina sequencing steps will reveal the imprint as a single strand break, we portray the imprint as a single strand break in the remainder of the text. In Figure 6A, we reasoned that upon chromatin sonication, the strand opposite to the nick will preferentially break at its vicinity. Library preparation using the Illumina kit (TruSeq Chip) uses an exonuclease and a DNA polymerase to process the ends to achieve efficient ligation prior to sequencing (Figure 6B). This method conserves the first nucleotide of the 5’ end of all sonicated fragments. Consequently, reads starting at the imprint are expected to be more frequent in the whole chromatin extract even prior to any chromatin immunoprecipitation. Furthermore, it is possible to count for each DNA strand (Watson and Crick) the number of 5′ ends at each position in the genome, and we will refer to this count as “5′ count” in the rest of the manuscript (see 5’ count analysis in Materials and Methods). A whole genome distribution of the 5’ count was established on the mat1M ∆2-3 (Figure 6C) and mat1P ∆2-3 and h90 strains (Figure 7B-C) using 130 nucleotide-long reads. 7 enriched regions were detected on the Watson strand (Figure 6C). 6 such regions were also detected on the Crick strand (Figure 6C). They delineate either repeated regions or regions exhibiting homology with the mitochondrial genome (Supplementary Table 1). The last enriched 5’ count is only found on the Watson strand and corresponds to the imprint site at mat1 (Figure 6C, in red).
–
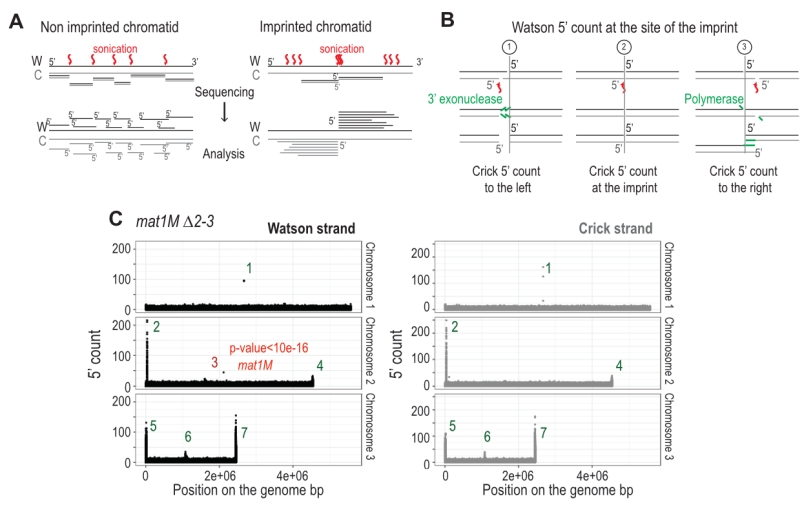 | FIGURE 6: The imprint site is detectable by high throughput sequencing on the Watson strand. (A) Schematic view of the result of the sonication of both chromatid types. Red arrows indicate sites of sonication breakage. The black strand is the Watson strand (W) and the gray the Crick (C). 5' of Watson are shown in black and those of Crick in gray. Lower panel indicates the different types of reads obtained after sequencing. (B) Schematic representation of the blunting of the end that is used during libraries preparation. The Crick and Watson 5' ends are preserved after treatment. The Crick strand can break at 3 positions compared to the imprint site: to the left, across or to the right. (C) Distribution of the 5' count per position in the genome on both strands. mat1M ∆2-3 stain was used for the analysis. p-value is calculated using a theoretical t-student distribution that fits on the distribution of the whole-genome observed 5' count. Regions that have a high 5' count are numerat-ed. |
We next focused on the 10-kb region surrounding the mat1 locus (Figure 7). The overall distribution of 5’ count is similar on the Watson and Crick strands, except for one nucleotide (in red) significantly over-represented on the Watson strand (Figure 7). The position in mat1M ∆2-3 and mat1P ∆2-3 corresponds to the imprinted position that is 3 nucleotides apart in M and P [22] (Figure 7A and 7B). As expected, both positions are enriched in 5′ count in the h90 background that contains a mixture of mat1M and mat1P alleles (Figure 7C). To confirm that the signal results from the imprint, we used the swi1∆ mutant strain that exhibits a low level of imprint. In this genetic background, no nucleotide shows any bias in 5′ count (Figure 7D).
–
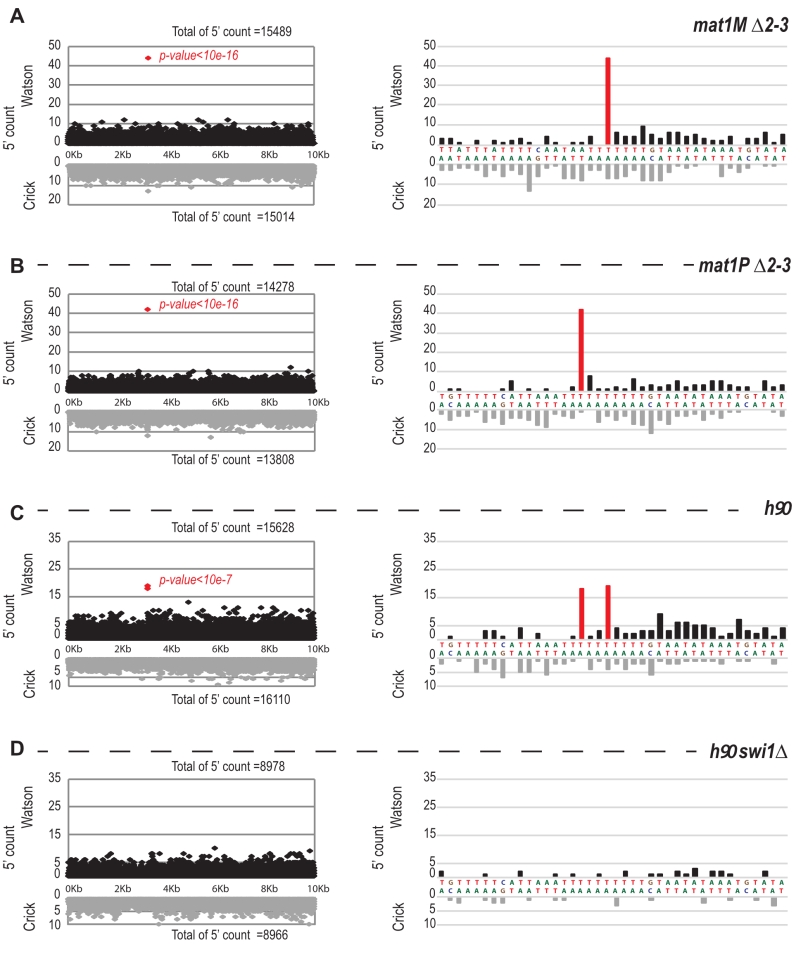 | FIGURE 7: 5' count on the Watson strand is biased precisely at the site of the imprint. On the left: distribution of the 5' count of the Watson strand (black) and the Crick strand (gray) on the 10-kb region containing mat1. On the right: zoom on the imprint site. Red bars represent the 5' count enriched at the imprint sites. p-value is calculated using a theoretical t-student distribution that fits on the distribution of the whole-genome observed 5' count. (A) The sequencing of mat1M ∆2-3 strain is represented, (B) mat1P ∆2-3 strain, (C) h90 strain and (D) h90 swi1∆ strain. ABCD) The mat1M ∆2-3, mat1P ∆2-3 and h90 P genomes are used, respectively. |
Because the bias in 5’ count is only visible on the imprinted strand, we conclude that sonication does not preferentially break the phosphate bond facing the nick. Therefore, we searched on the Crick strand for a region with a strong 5’ count statistical bias. Using a sliding window with different window sizes, we calculated the p-value in the 10-kb region surrounding mat1 (Figure 8). As expected, the window of one nucleotide at the site of the imprint has the lowest p-value on the Watson strand (Figure 8A and 8B). On the Crick strand, the most significant p-value is contained within a window of 17 to 23 nucleotides facing the imprint (Figure 8C and 8D). This corresponds to the molecular signature of a confined and persistent single-strand break upon chromatin sonication: A unique nucleotide on one strand and a larger region on the opposite strand (Figure 8E and 8F).
–
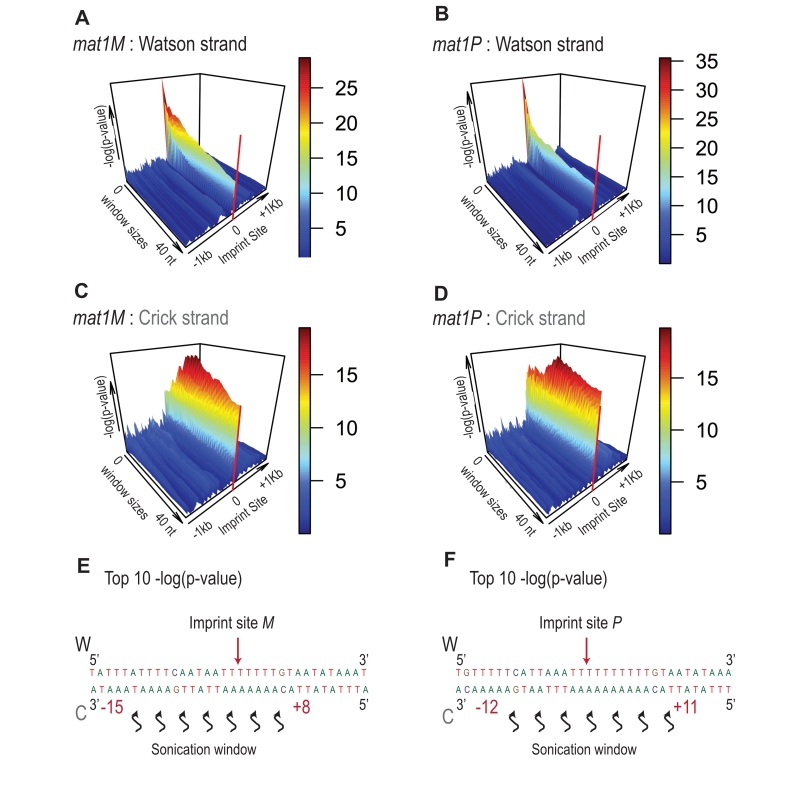 | FIGURE 8: Signature of sonication across the imprint. Statistical analysis of the 5' count in the mat1 region. -log(p-value) of 5' count as a function of the window size and genome position is represented. The -log(p-value) scale is indicated on the right of the graphics. The p-value is calculated using theoretical t-student distributions that fit with the distribution of 5' count observe in the 10-kb region that contains mat1. (A) A sliding window (+/- 1 kb around the imprint site) with step size one and varying window size (1 to 40 nt) is applied when calculating the -log(p-value) of the 5' count of the Watson strand shown. The sequencing of a mat1M ∆2-3 strain is used. (B) Same as in (A) for the Crick strand. (C) Window sizes and window positions compared to imprint site for the top 10 -log(p-value) are indicated. (D, E and F) Same as in (A, B and C), respectively for the sequencing of a mat1P ∆2-3 strain. |
Immunoprecipitation of the imprinted chromatid
To determine whether Lsd1, Lsd2, and Sap1 interact with the imprinted chromatid and on the right or left of the imprint we used the molecular signature defined above. Figure 9A shows the expected distributions of enriched 5’ count upon immunoprecipitation. Since Lsd1/2 and Sap1 bind the repeated mat cassettes, we circumvented the alignment problem by performing the experiment in the donor less strains. For Lsd1, the enrichment of the 5′ count at the imprint on the Watson and Crick strands (Figure 9B) at mat1M exhibits the molecular signature defined above (Figure 9C). This result shows that Lsd1 interacts on both sides or directly with the imprinted chromatid. The additional spreading of the 5’ count on both DNA strands indicates that Lsd1 also interacts with the chromatid in the absence of imprint (Figure 9B). A similar 5’ count distribution is observed for Lsd1 at mat1P (Figure 9D and 9E). Lsd2 and Sap1 exhibit a similar pattern at mat1M as well, while their interaction is weaker at mat1P (Figure 9D and 9E). Altogether, our results indicate that Lsd1, Lsd2 and Sap1 interact on both sides of the imprint but also with mat1 regardless of the imprint. Intriguingly, the interaction at mat1P is weaker for Lsd2 and Sap1.
–
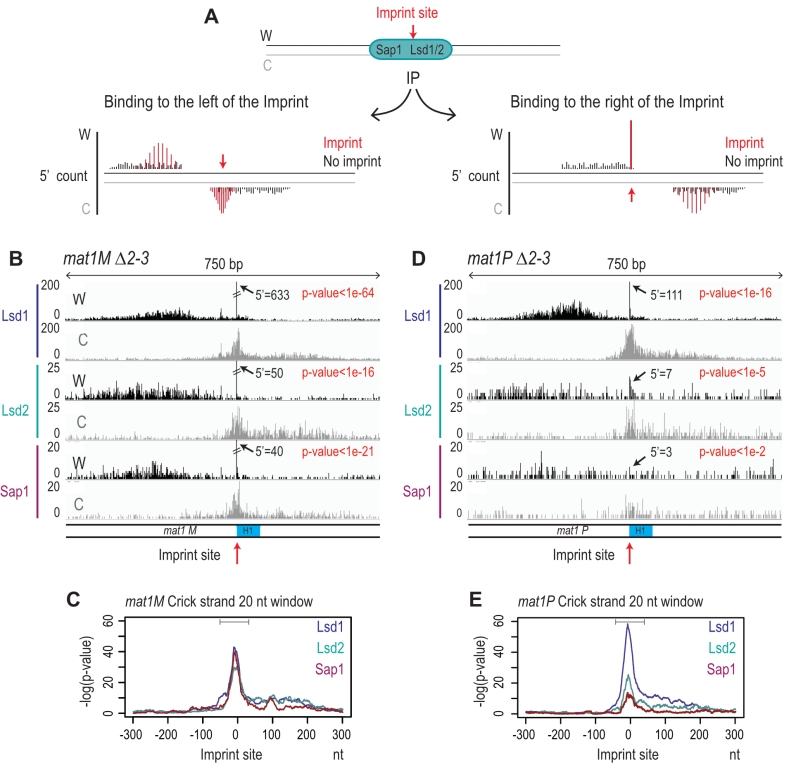 | FIGURE 9: Lsd1/2 and Sap1 immunoprecipitate chromatids on both sides of the imprint. Result of the 5' count analysis of the ChIP data presented in Figure 3C. (A) Schematic representation of the theoretical 5' count distribution of Lsd1/2 and Sap1. In black, the 5' count of a non-imprinted chromatid and in red that of an imprinted chromatid. (B) Distribution of the 5' count of the Lsd1/2 and Sap1 ChIPs for the Watson strand (black) and Crick strand (gray) in the mat1M ∆2-3 background. The 5' count at the imprint site is indicated. The p-value is calculated using theoretical t-student distributions that fit with the distribution of 5' count observed in the 10-kb region that contains mat1. (C) Analysis of the 5' count bias on the crick strand using a 20 nt window in a mat1M ∆2-3 strains for the IPs of Lsd1-2 and Sap1. A sliding window of 20 nt with set size one is used in a 10 kb region around mat1 is applied. Gray line indicates the bias of the crick across the imprint. (D) Same as in (B) in a mat1P ∆2-3 strain. (E) Same as in (C) in a mat1P ∆2-3 strain. |
DISCUSSION
In this work, we propose a molecular definition of the imprint upon sonication and genomic sequencing. Our computational analysis demonstrates that a site- and strand-specific persistent imprint/nick can be unambiguously mapped. The computation of the 5’ count reveals a unique accumulation at the site of the imprint on the Watson strand, whereas the opposite Crick strand is mechanically broken by sonication within a window of 20 nucleotides, thus yielding a more diffuse accumulation of 5’ ends. This molecular signature is consistent with the free rotation of the Crick strand that generates a population of molecules with weakened A-T rich Watson-Crick pairing on both sides of the nick and makes the resulting single-strand in this window highly prone to breakage. However, we cannot rule out that the proteins preventing the imprint from being repaired contribute as well to the pattern of shearing of the Crick strand.
–
We also provide data for the chromosomal enrichment at the genome wide level of four proteins, Lsd1, Lsd2, Sap1, and Abp1. Abp1 is strongly interacting with the sequences near mat1, but a function in replication fork pausing and imprinting was ruled out. The M-specific enrichment of Abp1 next to the Mc binding site located in the swi2 gene reinforces the role of Swi2 in directionality [42][43].
–
In mammals, Lsd1 plays an important role in gene expression by establishing and maintaining proper epigenetic marks. Several layers control the chromosomal targeting of Lsd1 for positive or negative transcription regulation. CoREST targets the Lsd1 complex to H3K4 [50], whereas the human androgen receptor targets Lsd1 to H3K9 [51]. The plant homeodomain finger protein BHC80 and the HMG protein Braf35 are part of the core complex Lsd1-CoREST (reviewed in [52]). BHC80 participates to the reading of histone marks [53] and Braf35 [54] is a structural DNA-binding protein.
–
In fission yeast, several pieces of evidence support the specific demethylation of H3K9 by Lsd1 at many promoters and boundary elements, but its role on H3K4 was not established [29][55]. Lsd1 is part of a complex including Lsd2 and Phf1/Phf2, two plant homeodomain finger proteins [53], and Lsd1 and Lsd2 are both fused to an HMG domain in fungi [56]. Taken together, the protein composition of the Lsd1/2 core complex is quite similar in fission yeast and mammals and the main functional difference resides in the absence of H3K4 demethylation. Because Sap1 interacts with the Lsd1/2 complex [29] and that Sap1 and Lsd1/2 complex are overlapping at several locations within the genome (see Supplemental Figure 1A) and notably at highly expressed genes (see Supplemental Figure 1B-E), we propose that Sap1 participates in the targeting of the Lsd1/2 complex to prevent H3K9 methylation at key promoters and therefore define heterochromatin boundaries [29][57].
–
The situation is more complicated at the mating type locus, where the methylation state of H3K9 is not involved in the early mating type switching steps. At this locus, the presence or absence of the sole H3K9 methyltransferase Clr4 does not rescue the mating type switching defect observed when lsd1 is mutated [27]. Although mat1P and mat1M exhibit no sequence homology, a remarkable finding is that Lsd1 and Lsd2 are enriched in both alleles on sequences that are necessary to promote replication fork pauses at MPS1 on the chromosome, and sufficient on a plasmid system [19]. Interestingly, when P and M sequences are present at the silent mat2P and mat3M loci, Lsd1 and Lsd2 are not detected (Supplemental Figure S2), consistent with the absence of replication fork pause at these loci [19]. In addition, the absence of H3K9 methylation (clr4 mutant) is not sufficient to trigger recruitment of Lsd1/2 at the silent mat2/3 loci (data not shown). We propose that the Phf1/Phf2 histone code readers participate to the targeting of Lsd1/2 at mat1 and probably at the other transcriptionally active loci [53].
–
By using the molecular signature of the imprint, we provide strong evidence that the Lsd1/2 complex is interacting at the imprint site, prior and after imprint formation. The same conclusion was reached by showing that Lsd1/2 is working upstream of Swi1/3 and is still enriched at mat1 in the absence of an imprint [27]. The two Sap1 binding sites on either side of the imprint (SS13 and SAS1) overlap with regions that are dispensable for both replication fork pausing and Lsd1/2 initial recruitment, but are essential for imprint formation or maintenance, at least when mat1 contains the M allele. When mat1 contains the P allele, only the SAS1 site remains, since no enrichment for Sap1 could be detected in the P sequences. This result further indicates that the processes required for replication fork pause and imprinting formation/protection are not identical in M and P cells. One possibility would be that Sap1 is either modified and not efficiently recognized during the immunoprecipitation or replaced by another protein in P cells.
–
The simplest model accounting for our results is that the Lsd1/2 complex is epigenetically recruited to mat1 independently of Sap1 to promote replication fork pausing at MPS1 (Figure 10, No Imprint). Following replication fork restart and imprinting, Sap1 and Lsd1/2 form a chromatin structure, the imprintosome, that protects the imprint from being repaired (Figure 10, Imprint). We propose that Lsd1/2 controls replication fork pausing at mat1 on both the imprinted and the non-imprinted chromatids, thus allowing the fork to restart in the same environment at each round of DNA replication and produce persistently and efficiently the imprint.
–
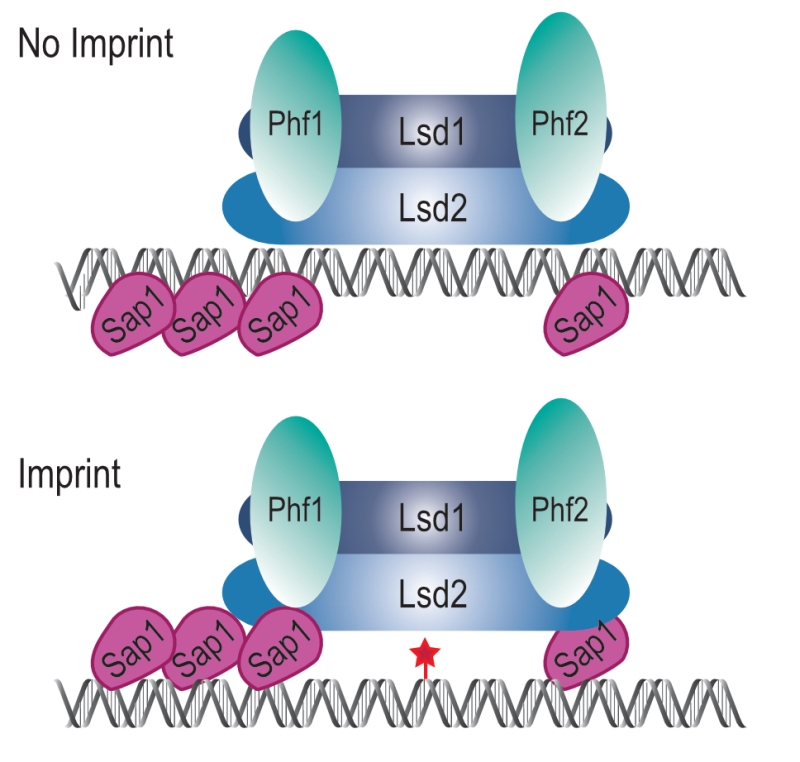 | FIGURE 10: The imprintosome. Schematic view of the model of binding of Lsd1/2 and Sap1 at mat1. Lsd1/2 complex is represented in blue with Phf1/2 in green. Sap1 is represented in pink. The imprint is represented by a red star. Upper panel, Lsd1/2 complex is recruited on the imprint site on the chromatid with no imprint independently of Sap1. Lower panel, when the imprint is formed Sap1 stabilizes the Lsd1/2 complex. |
MATERIALS AND METHODS
Fission yeast strains and genetic procedures
The S. pombe strains used in this study are listed in Supplemental Table S3. Standard media (YES) and genetic protocols for fission yeast were used [58].
–
2D Gel Analysis
2D gel analysis of replication intermediates was carried out as described previously [59]. DNA was prepared and digested in agarose plugs with the indicated restriction enzymes [12]. Enriched fractions for replication intermediates were obtained using BND cellulose columns (SIGMA). Gels were blotted onto Hybond-N+ nylon membrane. The probes were labeled with alpha-32P dCTP and the blot analyzed on a PhosphoImager and quantified with the ImageQuant software (pause/ arc + pause in percentage) (GE Healthcare Life Sciences). Sequences of primers used to produce the probe are indicated in Table 2.
–
Gel shift assay
Production of Sap1-6His by E. coli was induced with 1 mM IPTG as previously described [32]. Sap1-6His was purified using column of Ni-NTA His-tag from Qiagen according to the manufacturer’s instructions. Sequences used are listed in Table 2. 1.50 ng of a given oligonucleotide was labeled with gamma-32P with T4 polynucleotide Kinase (NEB) and incubated with its cold complementary sequence. 1 ng of the resulting double stranded probe was incubated with 5 µg of total protein extract or with 40 ng of purified Sap1-6His [44]. Gel shift was performed as previously described [60].
–
Chromatin Immunoprecipitation (ChIP)
ChIP-sequencing was performed as previously described [35] with the following modifications: fixation for ChIP of Lsd1 and Lsd2 was carried out at 25°C for 20 min with 1% of paraformaldehyde (PFA) and fixation for ChIP of Sap1 and Abp1 was performed at 18°C for 15 min with 3% of PFA. For Lsd1 and Lsd2, an Anti-Myc-Tag mAb-magnetic Beads (M047-11 MBL) was used. For Abp1 and Sap1, Dynabeads Protein G 10007D (Invitrogen) coupled with Tap Tag antibody CAB1001 (Pierce Thermo) or with polyclonal serum against the native Sap1[53], respectively, were used. For the ChIP followed by qPCR analysis, 3×108 cells were used with one tenth the amount of reagents.
–
quantitative PCR (q-PCR)
qPCR was performed using a master mix (Eurobio Green qPCR Mix Lo-Rox) on the qTower Analytik Jena AG A6 machine. Each value is the mean of technical triplicates. The enrichment was obtained using the ∆∆Cq normalization method using the value of a pair of primers localized 3 kb away from mat1 and the WCE values. The list of primer sequences is in Supplemental Table S4. Experiments were performed 3-times and unpaired t-test were used to calculate the p values.
–
Illumina Sequencing of DNA
Libraries were prepared using TruSeq ChIP Library Preparation kit following the manufacturer’s instructions (Illumina). Libraries are checked for concentration and quality on DNA chips with a Bioanalyser (Agilent). The libraries were quantified by fluorimetric measurements with the Qubit® dsDNA HS Assay Kit (ThermoFisher). 65-bp and 130-bp Single Read sequences were generated on the Hiseq2500 sequencer according to manufacturer’s instructions (Illumina). Library statistics are provided in Supplemental Table S5.
–
Illumina Read Processing and Alignment of DNA Libraries
Illumina reads were aligned to the S. pombe genome assembly ASM294v2.23 using Bowtie v2.1.0 [61] with default parameters and multi-mapper reads randomly distributed. Using BamTools [62], read counts were normalized to reads per million per kilo base (RPKM), using the total library size and a bin size of 1 kb. For the analysis of multi-mapper reads, we filtered aligned read files with samtools [63] using the parameter -q 10.
–
ChIP-Sequencing Analysis
ChIP peaks versus appropriate inputs (whole-cell extracts) were called using MACS v2.1.0 [64]. Statistic validation of peaks was performed using the IDR method with ENCODE recommendations [65]. Genome browser tracks and meta-analysis were created using enrichment (IP reads per million (RPM) / input RPM) of indicated sequencing. Intersection of lists of peaks were calculated with BEDTools v.2.17.02 [66] (parameters: -u). For the transcriptional analysis 2 groups of 108 genes were defined and intersected with the list of peaks shared by Lsd1/2 and Sap1. The first group contains the 108 most present transcripts and the 108 less present transcripts using the normalized data from [67].
–
5′ count analysis
We used a script to extract the beginning of reads that map on the Watson or Crick strands. Enrichments of 5′ count were obtained using the total library size. p-values were obtained using the negative binomial distribution fitting function in R.
–
All scripts used for the analysis and outputs are available upon request.
References
- ""http://www.ncbi.nlm.nih.gov/pubmed/14877267070512
- M. Kelly, J. Burke, M. Smith, A. Klar, and D. Beach, "Four mating-type genes control sexual differentiation in the fission yeast.", The EMBO journal, 1988. http://www.ncbi.nlm.nih.gov/pubmed/2900761
- D.H. Beach, "Cell type switching by DNA transposition in fission yeast", Nature, vol. 305, pp. 682-688, 1983. http://dx.doi.org/10.1038/305682a0
- R. Egel, and H. Gutz, "Gene activation by copy transposition in mating-type switching of a homothallic fission yeast", Current Genetics, vol. 3, pp. 5-12, 1981. http://dx.doi.org/10.1007/BF00419574
- R. Egel, "Two tightly linked silent cassettes in the mating-type region of Schizosaccharomyces pombe", Current Genetics, vol. 8, pp. 199-203, 1984. http://dx.doi.org/10.1007/BF00417816
- G. Thon, and J. Verhein-Hansen, "Four chromo-domain proteins of Schizosaccharomyces pombe differentially repress transcription at various chromosomal locations.", Genetics, 2000. http://www.ncbi.nlm.nih.gov/pubmed/10835380
- G. Thon, P. Bjerling, C.M. Bünner, and J. Verhein-Hansen, "Expression-state boundaries in the mating-type region of fission yeast.", Genetics, 2002. http://www.ncbi.nlm.nih.gov/pubmed/12072458
- K. Noma, C.D. Allis, and S.I.S. Grewal, "Transitions in Distinct Histone H3 Methylation Patterns at the Heterochromatin Domain Boundaries", Science, vol. 293, pp. 1150-1155, 2001. http://dx.doi.org/10.1126/science.1064150
- S.I. Grewal, and A.J. Klar, "A recombinationally repressed region between mat2 and mat3 loci shares homology to centromeric repeats and regulates directionality of mating-type switching in fission yeast.", Genetics, 1997. http://www.ncbi.nlm.nih.gov/pubmed/9258669
- B. Arcangioli, "Fission yeast switches mating type by a replication-recombination coupled process", The EMBO Journal, vol. 19, pp. 1389-1396, 2000. http://dx.doi.org/10.1093/emboj/19.6.1389
- A. Kaykov, A.M. Holmes, and B. Arcangioli, "Formation, maintenance and consequences of the imprint at the mating-type locus in fission yeast", The EMBO Journal, vol. 23, pp. 930-938, 2004. http://dx.doi.org/10.1038/sj.emboj.7600099
- O. Fleck, C. Rudolph, A. Albrecht, A. Lorentz, P. Schär, and H. Schmidt, "The mutator gene swi8 effects specific mutations in the mating-type region of Schizosaccharomyces pombe.", Genetics, 1994. http://www.ncbi.nlm.nih.gov/pubmed/7851760
- J.Z. Dalgaard, and A.J. Klar, "Does S. pombe exploit the intrinsic asymmetry of DNA synthesis to imprint daughter cells for mating-type switching?", Trends in genetics : TIG, 2001. http://www.ncbi.nlm.nih.gov/pubmed/11226610
- B. Arcangioli, and G. Thon, "Mating-Type Cassettes: Structure, Switching and Silencing", The Molecular Biology of Schizosaccharomyces pombe, pp. 129-147, 2004. http://dx.doi.org/10.1007/978-3-662-10360-9_9
- A.J.S. Klar, K. Ishikawa, and S. Moore, "A Unique DNA Recombination Mechanism of the Mating/Cell-type Switching of Fission Yeasts: a Review", Microbiology Spectrum, vol. 2, 2014. http://dx.doi.org/10.1128/microbiolspec.MDNA3-0003-2014
- A.M. Holmes, A. Kaykov, and B. Arcangioli, "Molecular and Cellular Dissection of Mating-Type Switching Steps in Schizosaccharomyces pombe", Molecular and Cellular Biology, vol. 25, pp. 303-311, 2005. http://dx.doi.org/10.1128/MCB.25.1.303-311.2005
- J.Z. Dalgaard, and A.J.S. Klar, "Orientation of DNA replication establishes mating-type switching pattern in S. pombe", Nature, vol. 400, pp. 181-184, 1999. http://dx.doi.org/10.1038/22139
- S. Sayrac, S. Vengrova, E.L. Godfrey, and J.Z. Dalgaard, "Identification of a Novel Type of Spacer Element Required for Imprinting in Fission Yeast", PLoS Genetics, vol. 7, pp. e1001328, 2011. http://dx.doi.org/10.1371/journal.pgen.1001328
- A. Kaykov, and B. Arcangioli, "A Programmed Strand-Specific and Modified Nick in S. pombe Constitutes a Novel Type of Chromosomal Imprint", Current Biology, vol. 14, pp. 1924-1928, 2004. http://dx.doi.org/10.1016/j.cub.2004.10.026
- S. Vengrova, and J.Z. Dalgaard, "The wild‐type Schizosaccharomyces pombe mat1 imprint consists of two ribonucleotides", EMBO reports, vol. 7, pp. 59-65, 2006. http://dx.doi.org/10.1038/sj.embor.7400576
- O. Nielsen, and R. Egel, "Mapping the double-strand breaks at the mating-type locus in fission yeast by genomic sequencing.", The EMBO journal, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2714252
- G. Thon, and A.J. Klar, "Directionality of fission yeast mating-type interconversion is controlled by the location of the donor loci.", Genetics, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8375648
- A.J. Klar, and L.M. Miglio, "Initiation of meiotic recombination by double-strand DNA breaks in S. pombe.", Cell, 1986. http://www.ncbi.nlm.nih.gov/pubmed/3742597
- L. Roseaulin, Y. Yamada, Y. Tsutsui, P. Russell, H. Iwasaki, and B. Arcangioli, "Mus81 is essential for sister chromatid recombination at broken replication forks", The EMBO Journal, vol. 27, pp. 1378-1387, 2008. http://dx.doi.org/10.1038/emboj.2008.65
- U. Styrkársdóttir, R. Egel, and O. Nielsen, "The smt-0 mutation which abolishes mating-type switching in fission yeast is a deletion.", Current genetics, 1993. http://www.ncbi.nlm.nih.gov/pubmed/8431959
- A. Holmes, L. Roseaulin, C. Schurra, H. Waxin, S. Lambert, M. Zaratiegui, R. Martienssen, and B. Arcangioli, "Lsd1 and Lsd2 Control Programmed Replication Fork Pauses and Imprinting in Fission Yeast", Cell Reports, vol. 2, pp. 1513-1520, 2012. http://dx.doi.org/10.1016/j.celrep.2012.10.011
- J.Z. Dalgaard, and A.J. Klar, "swi1 and swi3 perform imprinting, pausing, and termination of DNA replication in S. pombe.", Cell, 2000. http://www.ncbi.nlm.nih.gov/pubmed/11030618
- F. Lan, M. Zaratiegui, J. Villén, M.W. Vaughn, A. Verdel, M. Huarte, Y. Shi, S.P. Gygi, D. Moazed, R.A. Martienssen, and Y. Shi, "S. pombe LSD1 Homologs Regulate Heterochromatin Propagation and Euchromatic Gene Transcription", Molecular Cell, vol. 26, pp. 89-101, 2007. http://dx.doi.org/10.1016/j.molcel.2007.02.023
- T.B. Nicholson, and T. Chen, "LSD1 demethylates histone and non-histone proteins.", Epigenetics, 2009. http://www.ncbi.nlm.nih.gov/pubmed/19395867
- E. Noguchi, C. Noguchi, W.H. McDonald, J.R. Yates, and P. Russell, "Swi1 and Swi3 Are Components of a Replication Fork Protection Complex in Fission Yeast", Molecular and Cellular Biology, vol. 24, pp. 8342-8355, 2004. http://dx.doi.org/10.1128/MCB.24.19.8342-8355.2004
- B. Arcangioli, T.D. Copeland, and A.J. Klar, "Sap1, a protein that binds to sequences required for mating-type switching, is essential for viability in Schizosaccharomyces pombe.", Molecular and cellular biology, 1994. http://www.ncbi.nlm.nih.gov/pubmed/8114737
- E. Mejía-Ramírez, A. Sánchez-Gorostiaga, D.B. Krimer, J.B. Schvartzman, and P. Hernández, "The Mating Type Switch-Activating Protein Sap1 Is Required for Replication Fork Arrest at the rRNA Genes of Fission Yeast", Molecular and Cellular Biology, vol. 25, pp. 8755-8761, 2005. http://dx.doi.org/10.1128/MCB.25.19.8755-8761.2005
- G. Krings, and D. Bastia, "Sap1p Binds to Ter1 at the Ribosomal DNA of Schizosaccharomyces pombe and Causes Polar Replication Fork Arrest", Journal of Biological Chemistry, vol. 280, pp. 39135-39142, 2005. http://dx.doi.org/10.1074/jbc.M508996200
- M. Zaratiegui, M.W. Vaughn, D.V. Irvine, D. Goto, S. Watt, J. Bähler, B. Arcangioli, and R.A. Martienssen, "CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR", Nature, vol. 469, pp. 112-115, 2010. http://dx.doi.org/10.1038/nature09608
- L. Guan, P. He, F. Yang, Y. Zhang, Y. Hu, J. Ding, Y. Hua, Y. Zhang, Q. Ye, J. Hu, T. Wang, C. Jin, and D. Kong, "Sap1 is a replication-initiation factor essential for the assembly of pre-replicative complex in the fission yeast Schizosaccharomyces pombe", Journal of Biological Chemistry, vol. 292, pp. 6056-6075, 2017. http://dx.doi.org/10.1074/jbc.M116.767806
- J.Z. Jacobs, J.D. Rosado-Lugo, S. Cranz-Mileva, K.M. Ciccaglione, V. Tournier, and M. Zaratiegui, "Arrested replication forks guide retrotransposon integration", Science, vol. 349, pp. 1549-1553, 2015. http://dx.doi.org/10.1126/science.aaa3810
- A. Hickey, C. Esnault, A. Majumdar, A.G. Chatterjee, J.R. Iben, P.G. McQueen, A.X. Yang, T. Mizuguchi, S.I.S. Grewal, and H.L. Levin, "Single-Nucleotide-Specific Targeting of the Tf1 Retrotransposon Promoted by the DNA-Binding Protein Sap1 of Schizosaccharomyces pombe", Genetics, vol. 201, pp. 905-924, 2015. http://dx.doi.org/10.1534/genetics.115.181602
- T. Mizuguchi, N. Taneja, E. Matsuda, J. Belton, P. FitzGerald, J. Dekker, and S.I.S. Grewal, "Shelterin components mediate genome reorganization in response to replication stress", Proceedings of the National Academy of Sciences, vol. 114, pp. 5479-5484, 2017. http://dx.doi.org/10.1073/pnas.1705527114
- R. de Lahondès, V. Ribes, and B. Arcangioli, "Fission Yeast Sap1 Protein Is Essential for Chromosome Stability", Eukaryotic Cell, vol. 2, pp. 910-921, 2003. http://dx.doi.org/10.1128/EC.2.5.910-921.2003
- L. Aguilar-Arnal, F. Marsellach, and F. Azorín, "The fission yeast homologue of CENP-B, Abp1, regulates directionality of mating-type switching", The EMBO Journal, vol. 27, pp. 1029-1038, 2008. http://dx.doi.org/10.1038/emboj.2008.53
- E. Matsuda, R. Sugioka-Sugiyama, T. Mizuguchi, S. Mehta, B. Cui, and S.I.S. Grewal, "A homolog of male sex-determining factor SRY cooperates with a transposon-derived CENP-B protein to control sex-specific directed recombination", Proceedings of the National Academy of Sciences, vol. 108, pp. 18754-18759, 2011. http://dx.doi.org/10.1073/pnas.1109988108
- C. Yu, M.J. Bonaduce, and A.J.S. Klar, "Going in the Right Direction: Mating-Type Switching ofSchizosaccharomyces pombeIs Controlled by Judicious Expression of Two Differentswi2Transcripts", Genetics, vol. 190, pp. 977-987, 2012. http://dx.doi.org/10.1534/genetics.111.137109
- B. Arcangioli, and A.J. Klar, "A novel switch-activating site (SAS1) and its cognate binding factor (SAP1) required for efficient mat1 switching in Schizosaccharomyces pombe.", The EMBO journal, 1991. http://www.ncbi.nlm.nih.gov/pubmed/1915277
- M. Gordon, D.G. Holt, A. Panigrahi, B.T. Wilhelm, H. Erdjument-Bromage, P. Tempst, J. Bähler, and B.R. Cairns, "Genome-Wide Dynamics of SAPHIRE, an Essential Complex for Gene Activation and Chromatin Boundaries", Molecular and Cellular Biology, vol. 27, pp. 4058-4069, 2007. http://dx.doi.org/10.1128/MCB.02044-06
- K. Noma, H.P. Cam, R.J. Maraia, and S.I. Grewal, "A Role for TFIIIC Transcription Factor Complex in Genome Organization", Cell, vol. 125, pp. 859-872, 2006. http://dx.doi.org/10.1016/j.cell.2006.04.028
- H.P. Cam, K. Noma, H. Ebina, H.L. Levin, and S.I.S. Grewal, "Host genome surveillance for retrotransposons by transposon-derived proteins", Nature, vol. 451, pp. 431-436, 2007. http://dx.doi.org/10.1038/nature06499
- S. Vengrova, and J.Z. Dalgaard, "RNase-sensitive DNA modification(s) initiates S. pombe mating-type switching", Genes & Development, vol. 18, pp. 794-804, 2004. http://dx.doi.org/10.1101/gad.289404
- B. Arcangioli, "A site- and strand-specific DNA break confers asymmetric switching potential in fission yeast", The EMBO Journal, vol. 17, pp. 4503-4510, 1998. http://dx.doi.org/10.1093/emboj/17.15.4503
- Y. Shi, F. Lan, C. Matson, P. Mulligan, J.R. Whetstine, P.A. Cole, R.A. Casero, and Y. Shi, "Histone Demethylation Mediated by the Nuclear Amine Oxidase Homolog LSD1", Cell, vol. 119, pp. 941-953, 2004. http://dx.doi.org/10.1016/j.cell.2004.12.012
- E. Metzger, M. Wissmann, N. Yin, J.M. Müller, R. Schneider, A.H.F.M. Peters, T. Günther, R. Buettner, and R. Schüle, "LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription", Nature, vol. 437, pp. 436-439, 2005. http://dx.doi.org/10.1038/nature04020
- K. Meier, and A. Brehm, "Chromatin regulation: How complex does it get?", Epigenetics, vol. 9, pp. 1485-1495, 2014. http://dx.doi.org/10.4161/15592294.2014.971580
- F. Lan, R.E. Collins, R. De Cegli, R. Alpatov, J.R. Horton, X. Shi, O. Gozani, X. Cheng, and Y. Shi, "Recognition of unmethylated histone H3 lysine 4 links BHC80 to LSD1-mediated gene repression", Nature, vol. 448, pp. 718-722, 2007. http://dx.doi.org/10.1038/nature06034
- M. Hakimi, D.A. Bochar, J. Chenoweth, W.S. Lane, G. Mandel, and R. Shiekhattar, "A core–BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes", Proceedings of the National Academy of Sciences, vol. 99, pp. 7420-7425, 2002. http://dx.doi.org/10.1073/pnas.112008599
- M. Opel, D. Lando, C. Bonilla, S.C. Trewick, A. Boukaba, J. Walfridsson, J. Cauwood, P.J. Werler, A.M. Carr, T. Kouzarides, N.V. Murzina, R.C. Allshire, K. Ekwall, and E.D. Laue, "Genome-Wide Studies of Histone Demethylation Catalysed by the Fission Yeast Homologues of Mammalian LSD1", PLoS ONE, vol. 2, pp. e386, 2007. http://dx.doi.org/10.1371/journal.pone.0000386
- E. Nicolas, M.G. Lee, M. Hakimi, H.P. Cam, S.I. Grewal, and R. Shiekhattar, "Fission Yeast Homologs of Human Histone H3 Lysine 4 Demethylase Regulate a Common Set of Genes with Diverse Functions", Journal of Biological Chemistry, vol. 281, pp. 35983-35988, 2006. http://dx.doi.org/10.1074/jbc.M606349200
- F. Li, M. Huarte, M. Zaratiegui, M.W. Vaughn, Y. Shi, R. Martienssen, and W.Z. Cande, "Lid2 Is Required for Coordinating H3K4 and H3K9 Methylation of Heterochromatin and Euchromatin", Cell, vol. 135, pp. 272-283, 2008. http://dx.doi.org/10.1016/j.cell.2008.08.036
- S. Moreno, A. Klar, and P. Nurse, "Molecular genetic analysis of fission yeast Schizosaccharomyces pombe.", Methods in enzymology, 1991. http://www.ncbi.nlm.nih.gov/pubmed/2005825
- B.J. Brewer, and W.L. Fangman, "A replication fork barrier at the 3′ end of yeast ribosomal RNA genes", Cell, vol. 55, pp. 637-643, 1988. http://dx.doi.org/10.1016/0092-8674(88)90222-X
- B. Arcangioli, and B. Lescure, "Identification of proteins involved in the regulation of yeast iso- 1-cytochrome C expression by oxygen.", The EMBO journal, 1985. http://www.ncbi.nlm.nih.gov/pubmed/2996882
- B. Langmead, C. Trapnell, M. Pop, and S.L. Salzberg, "Ultrafast and memory-efficient alignment of short DNA sequences to the human genome", Genome Biology, vol. 10, pp. R25, 2009. http://dx.doi.org/10.1186/gb-2009-10-3-r25
- D.W. Barnett, E.K. Garrison, A.R. Quinlan, M.P. Strömberg, and G.T. Marth, "BamTools: a C++ API and toolkit for analyzing and managing BAM files", Bioinformatics, vol. 27, pp. 1691-1692, 2011. http://dx.doi.org/10.1093/bioinformatics/btr174
- H. Li, and R. Durbin, "Fast and accurate short read alignment with Burrows–Wheeler transform", Bioinformatics, vol. 25, pp. 1754-1760, 2009. http://dx.doi.org/10.1093/bioinformatics/btp324
- Y. Zhang, T. Liu, C.A. Meyer, J. Eeckhoute, D.S. Johnson, B.E. Bernstein, C. Nusbaum, R.M. Myers, M. Brown, W. Li, and X.S. Liu, "Model-based Analysis of ChIP-Seq (MACS)", Genome Biology, vol. 9, 2008. http://dx.doi.org/10.1186/gb-2008-9-9-r137
- S.G. Landt, G.K. Marinov, A. Kundaje, P. Kheradpour, F. Pauli, S. Batzoglou, B.E. Bernstein, P. Bickel, J.B. Brown, P. Cayting, Y. Chen, G. DeSalvo, C. Epstein, K.I. Fisher-Aylor, G. Euskirchen, M. Gerstein, J. Gertz, A.J. Hartemink, M.M. Hoffman, V.R. Iyer, Y.L. Jung, S. Karmakar, M. Kellis, P.V. Kharchenko, Q. Li, T. Liu, X.S. Liu, L. Ma, A. Milosavljevic, R.M. Myers, P.J. Park, M.J. Pazin, M.D. Perry, D. Raha, T.E. Reddy, J. Rozowsky, N. Shoresh, A. Sidow, M. Slattery, J.A. Stamatoyannopoulos, M.Y. Tolstorukov, K.P. White, S. Xi, P.J. Farnham, J.D. Lieb, B.J. Wold, and M. Snyder, "ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia", Genome Research, vol. 22, pp. 1813-1831, 2012. http://dx.doi.org/10.1101/gr.136184.111
- A.R. Quinlan, and I.M. Hall, "BEDTools: a flexible suite of utilities for comparing genomic features", Bioinformatics, vol. 26, pp. 841-842, 2010. http://dx.doi.org/10.1093/bioinformatics/btq033
- S. Marguerat, A. Schmidt, S. Codlin, W. Chen, R. Aebersold, and J. Bähler, "Quantitative Analysis of Fission Yeast Transcriptomes and Proteomes in Proliferating and Quiescent Cells", Cell, vol. 151, pp. 671-683, 2012. http://dx.doi.org/10.1016/j.cell.2012.09.019
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
AUTHOR CONTRIBUTIONS
C.R performed experiments. C.P pre-pared the libraries for sequencing. H.W provided a purified Sap1 protein. C.R and B.J performed the computational analysis. C.R and B.A conceived experiments. C.R., S.G. and B.A. wrote the manuscript.
ACKNOWLEDGMENTS
This work has been supported by the Fondation ARC for Cancer Research and by the Fondation pour la Recherche Médicale (FRM) and the Pasteur Institute. We are dedicating this article to the memory of Amar Klar and Jacob Dalgaard, two great scientists who have investigated for several decades the epigenetic aspects of mating-type switching in fission yeast. Amar Klar passed away in March and Jacob Dalgaard in December. Our community has lost two insightful and keen members.
COPYRIGHT
© 2018

Molecular signature of the imprintosome complex at the mating-type locus in fission yeast by Raimondi et al. is licensed under a Creative Commons Attribution 4.0 International License.








