
Research Articles:
Microbial Cell, Vol. 1, No. 12, pp. 393 - 405; doi: 10.15698/mic2014.12.178
Rewiring yeast acetate metabolism through MPC1 loss of function leads to mitochondrial damage and decreases chronological lifespan
1 SYSBIO Centre for Systems Biology Milano, Italy.
2 Dipartimento di Biotecnologie e Bioscienze, Università di Milano-Bicocca, Piazza della Scienza 2, 20126 Milano, Italy.
Keywords: acetyl-CoA, chronological aging, Mcp1, mitochondria, pyruvate, Saccharomyces cerevisiae.
Received originally: 28/06/2014 Received in revised form: 21/10/2014
Accepted: 05/11/2014
Published: 18/11/2014
Correspondence:
Marina Vai, Dipartimento di Biotecnologie e Bioscienze, Università di Milano-Bicocca, Piazza della Scienza 2; 20126 Milano, Italy marina.vai@unimib.it
Conflict of interest statement:
The authors declare no conflict of interest.
Please cite this article as: Ivan Orlandi, Damiano Pellegrino Coppola and Marina Vai (2014). Rewiring yeast acetate metabolism through MPC1 loss of function leads to mitochondrial damage and decreases chronological lifespan. Microbial Cell 1(12): 393-405.
Abstract
During growth on fermentable substrates, such as glucose, pyruvate, which is the end-product of glycolysis, can be used to generate acetyl-CoA in the cytosol via acetaldehyde and acetate, or in mitochondria by direct oxidative decarboxylation. In the latter case, the mitochondrial pyruvate carrier (MPC) is responsible for pyruvate transport into mitochondrial matrix space. During chronological aging, yeast cells which lack the major structural subunit Mpc1 display a reduced lifespan accompanied by an age-dependent loss of autophagy. Here, we show that the impairment of pyruvate import into mitochondria linked to Mpc1 loss is compensated by a flux redirection of TCA cycle intermediates through the malic enzyme-dependent alternative route. In such a way, the TCA cycle operates in a “branched” fashion to generate pyruvate and is depleted of intermediates. Mutant cells cope with this depletion byincreasing the activity of glyoxylate cycle and of the pathway which provides the nucleocytosolic acetyl-CoA. Moreover, cellular respiration decreases and ROS accumulate in the mitochondria which, in turn, undergo severe damage. These acquired traits in concert with the reduced autophagy restrict cell survival of the mpc1∆ mutant during chronological aging. Conversely, the activation of the carnitine shuttle by supplying acetyl-CoA to the mitochondria is sufficient to abrogate the short-lived phenotype of the mutant.
INTRODUCTION
Aging of postmitotic quiescent mammalian cells has been modelled in the yeast Saccharomyces cerevisiae by its chronological lifespan (CLS) [1][2]. CLS represents the length of time a culture of nondividing cells remains viable in stationary phase: viability is assessed by the ability to resume growth upon return to rich medium [3]. Evidence to date indicates that chronological aging is intimately regulated by signaling pathways which sense nutrient availability, namely TORC1-Sch9 and Ras-PKA, and carbon metabolism [4][5]. In this context, emerging data on some metabolites and nutrient manipulation/dietary regimens which proved to modulate aging not only in yeast but also in evolutionary diverse organisms have opened up new opportunities for therapeutic interventions promoting healthy aging in humans [6][7]. In particular, among the main metabolic intermediates, acetyl-CoA is increasingly being acknowledged as an important regulator of longevity [8][9][10]. This metabolite is the activated form of acetate obtained via a thioester linkage with coenzyme A which cells use for macromolecule biosynthesis. Furthermore, in the mitochondria it is a crucial substrate for energy production since it fuels the TCA cycle and consequently the production of reducing equivalents which enter the electron transport chain and support the oxidative phosphorylation. In addition, acetyl-CoA also supplies the acetyl group for protein acetylation, a dynamic posttranslational modification which occurs on a wide range of substrates, including histones and many metabolic enzymes, thus connecting metabolism, epigenetics and transcriptional regulation [11][12][13][14]. Recently, by manipulating the major routes of acetyl-CoA formation in yeast and mammalian cells, it has been shown that the nucleocytosolic pool of acetyl-CoA acts as a dominant suppressor of cytoprotective autophagy during aging [8][10]. In line with this, during aging, histone hypoacetylation correlates with enhanced expression of ATG genes and induction of autophagy [15]. This is a degradative process which is crucial for the maintenance of cellular homeostasis by removing misfolded/damaged or “obsolete” proteins and organelles, including mitochondria. It becomes fundamental in nondividing cells where the intracellular damage cannot be “diluted” [16]. Nutrient depletion and inactivations of genes in the central nutrient signaling pathways are known inducers of autophagy [17].
–
In S.cerevisiae, the nucleocytosolic pool of acetyl-CoA is synthesized by the acetyl-CoA synthetase 2 (Acs2) by activation of acetate in an ATP-dependent reaction. This enzyme is known as the glycolytic isoform [18] and besides its role in carbon metabolism it is required for histone acetylation [19]. The mitochondrial acetyl-CoA pool is generated by the Acs1 (the gluconeogenic isoform) and by the acetyl-CoA hydrolase 1 (Ach1) which catalyzes the transfer of the CoASH moiety from succinyl-CoA to acetate [20]. Moreover, according to culture conditions, acetyl-CoA can be formed and utilized in different ways. During growth on fermentable substrates, such as glucose, it is generated from pyruvate. This compound is the end-product of glycolysis and is a key node in the branching point between respiratory metabolism and alcoholic fermentation as well as assimilatory and dissimilatory metabolic reactions [21]. At the branching point, it can follow three major fates (Fig. 1): (i) decarboxylation to acetaldehyde which generates acetyl-CoA by the pyruvate dehydrogenase (PDH) bypass; (ii) anaplerotic carboxylation to oxaloacetate and (iii) the direct oxidative decarboxylation to acetyl-CoA by the PDH complex, which is located in the mitochondrial matrix. Pyruvate can cross the outer mitochondrial membrane while the passage across the inner mitochondrial membrane requires the mitochondrial pyruvate carrier (MPC) [22][23]. This carrier effectively represents a link between cytosolic pyruvate metabolism and the TCA cycle. Loss of the major structural subunit Mpc1 results in defective mitochondrial pyruvate uptake [22] and, during chronological aging, in a short-lived phenotype accompanied by an age-dependent loss of autophagy [8].
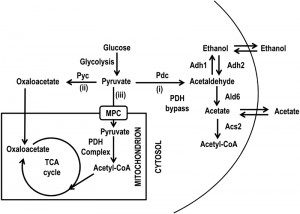 | FIGURE 1: Scheme of metabolic pathways allowing pyruvate utilization. The three pathways which originate from pyruvate after (i) decarboxylation to acetaldehyde, (ii) carboxylation to oxaloacetate and (iii) oxidative decarboxylation to acetyl-CoA are schematically shown. Acs, acetyl-CoA synthase; Adh, alcohol dehydrogenase; Ald, aldehyde dehydrogenase; MPC, mitochondrial pyruvate carrier; Pdc, pyruvate decarboxylase; PDH, pyruvate dehydrogenase; Pyc, pyruvate carboxylase. |
In this work we investigated the metabolic changes underlying MPC1 loss of function. We found that mpc1∆ cells make up for their impairment in mitochondrial pyruvate with a metabolic rewiring which involves several intermediates of the mitochondrially localized TCA cycle and the cytosolic glyoxylate shunt but ultimately results in a pro-aging process.
RESULTS AND DISCUSSION
Lack of Mpc1 is accompanied by an increase of Ald enzymatic activities
Since an impairment in the import of pyruvate into mitochondria linked to MPC1 deletion significantly restricted CLS (Fig. 2A) [8], we decided to analyze in more detail the metabolic changes underlying this short-lived phenotype. Initially, in the context of a standard CLS experiment [3], we measured the levels of some metabolites such as pyruvate, ethanol and acetate. These last two compounds are produced during glucose fermentation following decarboxylation of cytosolic pyruvate to acetaldehyde by pyruvate decarboxylase (Pdc) (Fig. 1). Only upon glucose depletion, does the diauxic shift occurs and yeast cells switch to a respiration-based metabolism of the fermentation C2 by-products. Finally, when these carbon/energy sources are fully exhausted, cells enter a quiescent stationary phase.At the diauxic shift, in the mpc1∆ culture the amount of ethanol and acetate was similar to that in the wild type (wt) culture (Fig. S1, 2B and C).Differently, during the post-diauxic phase, in the mutant the consumption of ethanol, which is re-introduced into the metabolism via its oxidation to acetate (Fig. 1), was not affected significantly compared to that in the wt (specific consumption rate, qEtOH, of 1.43 ± 0.04 mmol•g•DW-1•h-1 for the mutant and 1.12 ± 0.06 mmol•g•DW-1•h-1 for the wt) (Fig. S1 and 2B), while the acetatecontinued to accumulate in the medium (Fig. 2C). Such a prolonged secretion of acetate throughout the ethanol consumption phasesuggests that in the mpc1∆ mutant there is an imbalance between acetate production rate from acetaldehyde and its conversion rate into acetyl-CoA. In fact, the acetate transport relies on an active transport for the dissociated form of the acid (subjected to glucose repression) accompanied by passive/facilitated diffusion of the undissociated acid [24]. During the post-diauxic phase the pH of the medium is farbelow the pKa of acetic acid (4.75) [25] and according to the Henderson-Hasselbalch equation, acetic acid is substantially undissociated: 98.6 % at pH 2.9 (the value we measured at Day 3 after the diauxic shift). Consequently, in a condition where transmembrane diffusion strongly prevails over the active transport, the acetate export/import will take place according to the gradient between the intracellular and extracellular concentrations of acetate.
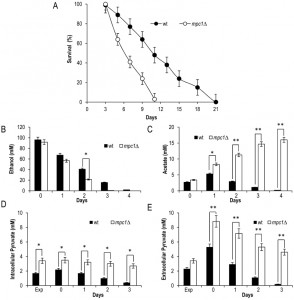 | FIGURE 2: MPC1 inactivation shortens CLS in concert with increased extracellular acetate and pyruvate. Wild type (wt) and mpc1Δ mutant cells were grown in minimal medium/2% glucose and the required supplements in excess (see Materials and Methods) and followed up to stationary phase. (A) CLS of wt and mpc1Δ mutant cells. At each time-point, survival was determined by colony-forming capacity. 72 h after the diauxic shift (Day 3) was considered the first age-point (see Materials and Methods). Day 0, diauxic shift. Data refer to mean values of three independent experiments. Standard deviations (SD) are indicated. Bar charts of extracellular ethanol (B) and acetate (C) concentrations at different time points after the diauxic shift (Day 0). In parallel, intracellular (D) and extracellular (E) pyruvate concentrations were measured. Exp, exponential growth phase. Data refer to mean values of three independent experiments. SD is indicated. Statistical significance as assessedby one-way ANOVA test is indicated (* P ≤ 0.05 and ** P ≤ 0.01). |
Concerning intracellular pyruvate, its concentration was higher in the mpc1∆ mutant compared to that in thewt not only in exponential phase, as already observed by [22], but also at/after the diauxic shift (Fig. 2D). This was also associated with an increase in the extracellular pyruvate (Fig. 2E) which reflects an overflow of pyruvate within the cytosol.Similar results were obtained when the experiments were also performed by growing the histidine-prototrophmpc1∆ mutant (mcp1∆::HIS3) in a histidine-supplemented medium as previously carried out for the wt (Fig. S2) indicating that the different composition of amino acids in the medium does not affect the results.
–
Afterwards, we measured the enzymatic activities of alcohol dehydrogenases (Adhs) catalysing the interconversion of acetaldehyde and ethanol [26] and of acetaldehyde dehydrogenases (Alds) which produce acetate by oxidizing the acetaldehyde generated from pyruvate during fermentation and that obtained during ethanol oxidation (Fig. 1). No significant difference was found between the wt and the mpc1∆ strain in the Adh activity levels in exponential phase (Fig. 3A), where the Adh1 isoenzyme is chiefly responsible for ethanol formation from acetaldehyde, consistent with the similar amounts of ethanol detected in both cultures (Fig. 2B). Similarly, at/after the diauxic-shift where the cytosolic Adh2 is the major ethanol oxidizer, Adh activities displayed no significant difference (Fig. 3A). On the contrary, during all the growthphases analyzed, Ald activity levels were higher in the mutant compared with the wt (Fig. 3B). In particular, a great increase was observed for Ald6 which is the major cytosolic isoform and is not glucose-repressed [27] compared with that of the mitochondrial counterparts Ald5 and Ald4 (Fig. 3C and D) indicating that the mutant exhibits an increased ability to generate acetate, especially the cytosolic one, which can be used as substrate to produce acetyl-CoA. Accordingly, in the mutant, the nucleocytosolic Acs2-mediated pathway is upregulated during chronological aging [8]. Moreover, an increased cytosolic acetate pool can also account for the extracellular acetate detected in the mpc1∆ culture (Fig. 2C) whose prolonged accumulation, however, indicates that the fluxtowards its formation exceeds its utilization. This takes place despite the upregulation of Acs2 enzymatic activity [8] suggesting that the enzyme and/or the flux downstream is/are working at maximum capacity in line with data which show that increase in Acs activity does not result in enhanced acetate utilization [28][29].
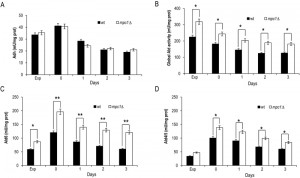 | FIGURE 3: In mpc1∆ cells the extracellular abundance of acetate correlates with enhanced Ald enzymatic activity. Bar charts of total Adh (A), total Ald (B), Ald6 (C) and Ald4/5 (D) enzymatic activities measured at the indicated time points for wt and mpc1Δ mutant cells grown as in Figure 2. Exp, exponential growth phase. Day 0, diauxic shift. Data refer to mean values determined in three independent experiments. SD is indicated. *P ≤ 0.05 and ** P ≤ 0.01. |
Lack of Mpc1 is accompanied by an increase of malic enzyme activity and a decrease in respiration
Starting from these results, we focused on the mitochondrially localized TCA cycle which can be fed with acetyl-CoA generated either from acetate or following oxidation of mitochondrial pyruvate. We measured the levels of citrate, succinate and malate which are intermediates of this cycle but also metabolic connections with the glyoxylate shunt. This is an anaplerotic device of the TCA cycle which allows the formation of C4 units from C2 units (acetate) by bypassing oxidative decarboxylation (Fig. 4A) [30]. At/after the diauxic shift, a clear global decrease was observed for all three intermediates in the mpc1∆ cells compared with the wt counterparts (Fig. 4B-D). This decrease was particularly marked for malate which can be used to generate pyruvate in the mitochondria for biosynthetic purposes. This reaction of oxidative decarboxylation is catalyzed by the mitochondrial malic enzyme encoded by MAE1 [31]. As shown in Fig. 4E, in the mpc1∆ mutant during the post-diauxic phase, the malic enzyme activity was doubled in comparison with the wt, suggesting that the impairment of pyruvate import into mitochondria linked to Mpc1 loss is compensated by a flux redirection through the Mae1-dependent alternative route. This can explain the severe growth defect observed by [22] when the mpc1∆ allele was combined with MAE1 deletion.
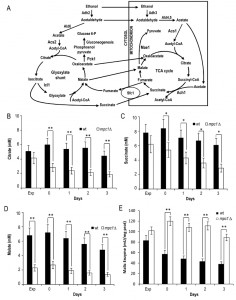 | FIGURE 4: Lack of Mpc1 results in low levels of TCA cycle intermediates and enhanced malic enzyme activity. (A) Scheme of the TCA cycle and of the glyoxylate shunt. Ach1, acetyl-CoA hydrolase 1; Acs, acetyl-CoA synthase; Adh, alcohol dehydrogenase; Ald, aldehyde dehydrogenase; Icl1, isocitrate lyase 1; Mae1, malic enzyme; Pck1,phosphoenolpyruvate carboxykinase 1; Sfc1, succinate-fumarate carrier. Wt and mpc1Δ cells were grown as in Figure 2 and at the indicated time points the concentrations of citrate (B), succinate (C) and malate (D) were measured. The bar chart of malic enzyme activity (E) is also reported. Exp, exponential growth phase. Day 0, diauxic shift. Data refer to mean values determined in three independent experiments. SD is indicated.* P ≤ 0.05 and ** P ≤ 0.01. |
Moreover, when cells switched to a respiration-based metabolism by using ethanol and acetate, the glyoxylate shunt becomes operative and begins replenishing the TCA cycle intermediates. In addition, during growth on C2 compounds, this shunt is the exclusive source of oxaloacetate which is the substrate of phosphoenolpyruvate carboxykinase (Pck1), the key enzyme of gluconeogenesis [32]. Measurements of the enzymatic activities of isocitrate lyase (Icl1), which is one of the unique enzymes of the glyoxylate shunt, and Pck1 indicated that these activities were higher in mpc1∆ cells compared with wt ones (Fig. 5A and B). Concomitantly, in mpc1∆ cells cellular respiration decreased (Fig. 5C). Icl1 is localized in the cytosol and from isocitrate it generates succinate and the name-giving metabolite glyoxylate which condenses with acetyl-CoA yielding malate. The last one can return to the mitochondria (Fig. 4A). Similarly, the major fate of cytosolic succinate is assumed to be its transfer into mitochondria [30]. Moreover, its transport by the Sfc1 carrier provides cytosolic fumarate for conversion to malate which can be used for gluconeogenesis [33]. Thus, taken together, these data indicate that in the mpc1∆ mutant, an increase in the glyoxylate shunt might represent an increase in metabolite feeding from the cytosol to support a mitochondrial impaired TCA cycle. In this context, the cytosol of the mutant can provide the metabolic environment required to fulfill the increased requirement of substrates for the glyoxylate shunt. In fact, the end-product of the Acs2 synthetase, which is increased in the mutant [8], is the nucleocytosolic acetyl-CoA. In the cytosol, this metabolite, following condensation with oxaloacetate, produces citrate which is then isomerized to isocitrate (the substrate of Icl1). In addition, the cytosol of the mutant might also be a suitable environment which can “promote” Pck1 enzymatic activity. In fact, Pck1 is acetylated by Esa1 and this acetylation is required for its enzymatic activity: an increase of Pck1 enzymatic activity is associated with an increase of the acetylated form of the enzyme [34][35]. Accumulating evidence indicates that the availability of acetyl-CoA, the donor substrate for acetylation, can be a metabolic input for the acetylation itself [36][37][38], so it is reasonable to hypothesize that changes of acetyl-CoA levels may also influence Esa1 activity.
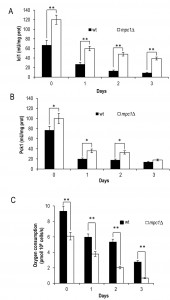 | FIGURE 5: MPC1 inactivation increases glyoxylate/gluconeo-genesis and reduces respiration during chronological aging. At the indicated time points Icl1 (A) and Pck1 (B) enzymatic activities of wt and mpc1Δ mutant cells were measured. In parallel, cellular respiration (C) was also monitored. Day 0, diauxic shift. Data refer to mean values determined in three independent experiments. SD is indicated. * P ≤ 0.05 and ** P ≤ 0.01. |
Carnitine restores chronological longevity of the mpc1∆ mutant
After the diauxic shift, a metabolic change from fermentation to respiration takesplace implying that energy metabolism relies on mitochondrial functionality. Since in the mpc1∆ cells we observed a decrease in respiration, we decided to analyze mitochondrial membrane potential and morphology by using the fluorescent dye, 3,3”-dihexyloxacarbocyanine iodide (DiOC6) [39]. In fact, mitochondrial morphology reflects the functional status of mitochondria and is regulated by the orchestrated balance of two opposing events: fission and fusion of mitochondria [40]. As shown in Fig. 6A, a typical tubular network was observed for wt cells whereas for the mutant fluorescent punctiform structures appeared at Day 2 after the diauxic shift. These structures are indicative of mitochondrial fragmentation and are linked to an elevated activity of the mitochondrial fission machinery [41]. In addition to an altered morphology, the mitochondria of the mutant displayed a time-dependent reduction in membrane potential and, at Day 4, did not accumulate DiOC6 (Fig.6A). Mitochondrial dysfunctions are intrinsically related to reactive oxygen species (ROS) of which superoxide anion is one of the most potentially harmful. This radical derives mainly from leakage of electrons from the respiratory chain and, among others, can target mitochondria with detrimental effects [42][43]. Chronologically aging mpc1∆ cells had a higher ROS content, measured as the superoxide-driven conversion of non-fluorescent dihydroethidium (DHE) into fluorescent ethidium (Eth), compared with that of the wt cells (Fig. 6B). Notably, culturing mpc1∆ cells in a carnitine-supplemented medium was sufficient to avoid this phenomenon (Fig. 6C). Moreover, oxygen consumption measurements indicated that in these cells cellular respiration increased (Fig. 6D).
 | FIGURE 6: Chronologically aging mpc1∆ cells display damaged mitochondria. (A) Representative images of wt and mpc1Δ cultures of Figure 2 stained with DiOC6 to visualize mitochondrial membranes. Morphologies of the mitochondria in late exponential phase (Late exp) are also shown.The same cultures were assessed for the presence of intracellular superoxide by conversion of non-fluorescentdihydroethidium into fluorescent ethidium (Eth). Summary graphs of the percentage of fluorescent/superoxide positive cells (% Eth) are reported (B). (C) Summary graphs of % Eth cells and (D) cellular respiration determined in wt and mpc1Δ cultures grown in minimal medium/2% glucose supplemented with carnitine (10 mg/L). Day 0, diauxic shift. For the determination of Eth cells, evaluation of about 1000 cells for each sample (three technical replicates) in three independent experiments was performed. SD is indicated. * P ≤ 0.05 and ** P ≤ 0.01. |
In S.cerevisiae, carnitine is involved in a process referred to as the carnitine shuttle which allows the transport of acetyl-CoA to the mitochondria. This transport system which is non-functional unless carnitine is supplied with the medium [44][45], involves the transfer of the acetyl moiety of acetyl-CoA to carnitine and the subsequent transport of the acetylcarnitine to the mitochondria. Here, a mitochondrial carnitine acetyltransferase catalyses the reverse reaction generating carnitine and acetyl-CoA which enters the TCA cycle [44][46].As shown in Fig. 7A-C, the supplemental carnitine did not significantly affect the levels of citrate, succinate and malate in the wt whilst this was not the case for the mpc1∆ mutant where the levels of all three intermediates increased and were restored to wt-like ones. No effect was observed on the malic enzyme activity which at/after the post diauxic shift in the mpc1∆ cells was still the double of that of the wt (Fig. 7D) suggesting that the presence of carnitine does not abolish the Mae1-dependent flux towards mitochondrial pyruvate generation. Concomitantly, in the mpc1∆ cells the enzymatic activities of Icl1 and Pck1 were reduced to the physiological levels measured in the wt (Fig. 7D and E). Thus, all this suggests that in the mpc1∆ mutant the activation of the carnitine shuttle can properly feed the TCA cycle by supplying acetyl-CoA to the mitochondria. Hence, the compensative metabolite feeding from the cytosol provided by the glyoxylate shunt seems to be no longer required.
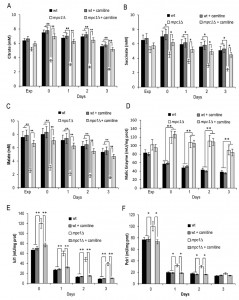 | FIGURE 7: Carnitine increases the levels of the TCA cycle intermediates in the mpc1∆ mutant. Wt and mpc1Δ cells were grown in minimal medium/2% glucose supplemented with carnitine (10 mg/L) and, at the indicated time points, the concentrations of citrate (A), succinate (B) and malate (C) were measured together with Mae1 (D), Icl1 (E) and Pck1 (F) enzymatic activities. Day 0, diauxic shift. Data refer to mean values determined in three independent experiments. SD is indicated. * P ≤ 0.05 and ** P ≤ 0.01. |
Moreover, following carnitine supplementation, during the post-diauxic phase no effect was observed on the ethanol consumption in both the wt and the mutant strains (Fig. 8A). Similarly, the acetate utilization in the wt was not affected, while in the mpc1∆ mutant its utilization was promoted (Fig. 8B). This indicates that in the latter the activation of the carnitine shuttle and the consequent acetyl-CoA transport to the mitochondria can result in an enhancement in the flux downstream from the acetate activation allowing acetate utilization. Finally, in the mpc1∆ mutant these metabolic changes matched the almost completely restored chronological longevity (Fig. 8C).
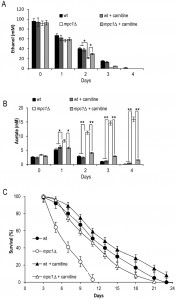 | FIGURE 8: Carnitine promotes acetate utilization in the mpc1∆ mutant in concert with increased CLS. Bar charts of extracellular ethanol (A) and acetate (B) concentrationsat different time points after the diauxic shift (Day 0) measured for cells grown as in Figure 7. (C) CLS of the same cells determined as in Figure 2. Data refer to mean values of three independent experiments. SD is indicated. * P ≤ 0.05 and ** P ≤ 0.01. |
In conclusion, these data collectively indicate that the lack of the Mpc1 transporter brings about a chain of metabolic events which, in order to counteract the decrease of the pyruvate supply in the mitochondria, by influencing the global acetyl-CoA metabolism ultimately restrict cell survival during chronological aging. In particular, after the diauxic shift when cells utilize the earlier produced ethanol/acetate and increase their respiration demand, one of the metabolic traits of the mpc1∆ mutant is a TCA cycle operating in a “branched” fashion with a propensity to shunt intermediates towards pyruvate generation via the malic enzyme. This kind of not-complete cyclic functioning of the TCA cycle by depleting it of intermediates influences not only the respiration, which is reducedin the mutant, but also might reduce mitochondrial acetyl-CoA pool. In fact, a TCA cycle characterized by low levels of intermediates (Fig. 4B-D) generates less succinyl-CoA. This is the substrate for the CoA-transferase’s reaction from succinyl-CoA to acetate, catalyzed by Ach1 in cells released from glucose repression [20]. In the mitochondria, this reaction allows the production of acetyl-CoA [8]. Moreover, the CoA-transferase’s reaction is using acetate as acceptor, which implies that Ach1 is also important for acetate detoxification and mitochondrial functionality during chronological aging [47]. Consequently, in a condition where acetate-generating activities of Ald4/Ald5 are increased, as it is the case in the mutant, a reduction in CoA-transferase’s enzymatic activity could play a causative role in promoting/enhancing the mitochondrial damage observed in the mutant.
–
Furthermore, since during the utilization of ethanol and acetate, the sole possible route for the net synthesis of C4 dicarboxylic acids for replenishing the TCA cycle of intermediates is the glyoxylate shunt, it follows that in the mpc1∆ cells this anaplerotic shunt is enhanced in order to keep the “branched” TCA cycle functioning. In turn, it follows that the pathway providing the cytosolic acetyl-CoA must be increased to support an enhanced glyoxylate demand. In line with this, the cytosolic Ald6 enzymatic activity is increased (Fig. 3C) and hyperactivation of the Acs2 activity has been detected [8]. Interestingly, this synthetase is responsible not only for supplying acetyl-CoA for carbon metabolism, but also for protein acetylation, particularly of histones [19]. In addition, it has been shown that during chronological aging, upregulation of Acs2 activity culminates in histone H3 hyperacetylation associated with transcriptional downregulation of several autophagy-essential ATG genes [48]. In this context, mpc1∆ cells display an age-dependent loss of autophagy [8]; this feature, given the reciprocal cross-talk between autophagy and mitochondria, can negatively affect the removal of the damaged mitochondria of the mutant and consequently contribute to its inability to maintain proper cellular homeostasis during the aging process. Notably, Ald6, which is responsible of generating cytosolic acetate, is degraded preferentially by autophagy [49] and the persistence of its enzymatic activity seems to be disadvantageous for the survival during nitrogen starvation [50]. Thus, mpc1∆ cells make up for their impairment in mitochondrial pyruvate with a metabolic rewiring in which the pro-aging outcome prevails.
MATERIALS AND METHODS
Yeast strains and growth conditions
The mcp1∆ mutant (mcp1∆::HIS3) was generated by PCR-based methods in a BY4741 background (MATa his3∆-1 leu2∆-0 met15∆-0 ura3∆-0) andthe accuracy of gene replacement was verified by PCR with flanking and internal primers. At least two different clones were tested for any experiment. Yeast cells were grown in batches at 30°Cin minimal medium (Difco Yeast Nitrogen Base without amino acids, 6.7 g/L) with 2% glucose andthe required supplements added in excess to a final concentration of 200 mg/L, except for leucine at 500 mg/L to avoid auxotrophy starvation [51][52]. L-carnitine (Sigma) was supplemented to a concentration of 10 mg/L. Strains were inoculated at the same cellular density (culture volume no more than 20% of the flask volume) and growth was monitored by determining cell number using a Coulter Counter-Particle Count and Size Analyser, as described [53]. Duplication times (Td) were obtained by linear regression of the cell number increase over time on a semi-logarithmic plot.
–
CLS determination
Survival experiments in expired medium were performed on cells grown in minimal medium/2% glucose and the required supplements as described above. During growth, cell number and extracellular glucose, ethanol and acetic acid were measured in order to define the growth profile (exponential phase, diauxic shift, post-diauxic phase and stationary phase) of the culture (Fig. S1). Cell survival was monitored by harvesting aliquots of cells starting with 72 h (Day 3, first age-point) after the diauxic shift (Day 0). CLS was measured according to [51] by counting colony-forming units (CFUs) every 2-3 days. The number of CFUs on Day 3 was considered the initial survival (100%).
–
Metabolite measurements and enzymatic assays
At designated time points, aliquots of the yeast cultures were centrifuged and both pellets (washed twice) and supernatants were frozen at −80°C until used. Rapid sampling for intracellular metabolite measurements was performed according to the leakage-free cold methanol quenching method developed by [54] in which pure methanol at ≤ -40°C and a ratio of cell culture to quenching solvent of 1:5 (final methanol concentration ≥ 83%) were used. Metabolites from the cell pellets were extracted in 5 ml of a solution of 75% (v/v) boiling absolute ethanol containing 0.25 M Hepes, pH 7.5, as described in [55]. The concentrations of glucose, ethanol, acetate, pyruvate, citrate, succinate and malate were determined using enzymatic assays (K-HKGLU, K-ETOH, K-ACET, K-PYRUV, K-SUCC, K-CITR and K-LMALR kits from Megazyme). Ethanol specific consumption rate (qEtOH), expressed in mmol•g•DW -1•h-1, was calculated from measured cell dry weights (DWs) and extracellular ethanol concentrations. DW was measured as described [56].
–
All the enzymatic activities were assayed immediately after preparation of cell-free extracts. Cells were resuspended in 100 mM potassium phosphate buffer, pH 7.5, containing 2 mM MgCl2 and 1 mM dithiothreitol and broken with acid-washed glass beads by shaking on a vortex for several cycles interspersed with cooling on ice. The activities of cytosolic and mitochondrial aldehyde dehydrogenase (Ald) were measured as described by [57], of alcohol dehydrogenase (Adh) according to [58], of phosphoenolpyruvate carboxykinase (Pck1) and isocitrate lyase (Icl1) as in [34]. Malic enzyme activities were determined according to [31] with either 0.4 mM NAD+ or NADP+ as the redox cofactor. The enzymatic activity was measured in the decarboxylation direction to avoid interference with pyruvate decarboxylase and Adh. Total protein concentration was estimated using the BCATM Protein Assay Kit (Pierce).
–
Oxygen consumption and fluorescence microscopy
The basal oxygen consumption of intact cells was measured at 30°C using a ‘Clark-type’ oxygen electrode in a thermostatically controlled chamber (Oxygraph System, Hansatech Instruments, Norfolk, UK) as previously reported [25]. Data were recorded at sampling intervals of 1 s (Oxygraph Plus software, Hansatech Instruments, Norfolk, UK). All assays were conducted in biological triplicate.
–
ROS were detected with dihydroethidium (DHE, Sigma) according to [59]. The mitochondrial membrane potential was assessed by staining with DiOC6 (Molecular Probes, Invitrogen), according to [39]; cells were also counterstained with propidium iodide to discriminate between live and dead cells. A Nikon Eclipse E600 fluorescence microscope equipped with a Leica DC 350F ccd camera was used. Digital images were acquired using FW4000 software (Leica).
–
Statistical analysis of data
All values are presented as the mean of three independent experiments with the corresponding Standard Deviation (SD). Three technical replicates were analyzed in each independent experiments. Statistical significance was assessed by one-way ANOVA test. P value of ≤ 0.05 was considered statistically significant.
References
- V.D. Longo, and B.K. Kennedy, "Sirtuins in Aging and Age-Related Disease", Cell, vol. 126, pp. 257-268, 2006. http://dx.doi.org/10.1016/j.cell.2006.07.002
- M. MacLean, N. Harris, and P.W. Piper, "Chronological lifespan of stationary phase yeast cells; a model for investigating the factors that might influence the ageing of postmitotic tissues in higher organisms", Yeast, vol. 18, pp. 499-509, 2001. http://dx.doi.org/10.1002/yea.701
- P. Fabrizio, and V.D. Longo, "The chronological life span of Saccharomyces cerevisiae", Aging Cell, vol. 2, pp. 73-81, 2003. http://dx.doi.org/10.1046/j.1474-9728.2003.00033.x
- L. Fontana, L. Partridge, and V.D. Longo, "Extending Healthy Life Span—From Yeast to Humans", Science, vol. 328, pp. 321-326, 2010. http://dx.doi.org/10.1126/science.1172539
- E. Swinnen, R. Ghillebert, T. Wilms, and J. Winderickx, "Molecular mechanisms linking the evolutionary conserved TORC1-Sch9 nutrient signalling branch to lifespan regulation inSaccharomyces cerevisiae", FEMS Yeast Research, vol. 14, pp. 17-32, 2013. http://dx.doi.org/10.1111/1567-1364.12097
- R.H. Houtkooper, R.W. Williams, and J. Auwerx, "Metabolic Networks of Longevity", Cell, vol. 142, pp. 9-14, 2010. http://dx.doi.org/10.1016/j.cell.2010.06.029
- R. de Cabo, D. Carmona-Gutierrez, M. Bernier, M. Hall, and F. Madeo, "The Search for Antiaging Interventions: From Elixirs to Fasting Regimens", Cell, vol. 157, pp. 1515-1526, 2014. http://dx.doi.org/10.1016/j.cell.2014.05.031
- T. Eisenberg, S. Schroeder, A. Andryushkova, T. Pendl, V. Küttner, A. Bhukel, G. Mariño, F. Pietrocola, A. Harger, A. Zimmermann, T. Moustafa, A. Sprenger, E. Jany, S. Büttner, D. Carmona-Gutierrez, C. Ruckenstuhl, J. Ring, W. Reichelt, K. Schimmel, T. Leeb, C. Moser, S. Schatz, L. Kamolz, C. Magnes, F. Sinner, S. Sedej, K. Fröhlich, G. Juhasz, T. Pieber, J. Dengjel, S.J. Sigrist, G. Kroemer, and F. Madeo, "Nucleocytosolic Depletion of the Energy Metabolite Acetyl-Coenzyme A Stimulates Autophagy and Prolongs Lifespan", Cell Metabolism, vol. 19, pp. 431-444, 2014. http://dx.doi.org/10.1016/j.cmet.2014.02.010
- R. Friis, J. Glaves, T. Huan, L. Li, B. Sykes, and M. Schultz, "Rewiring AMPK and Mitochondrial Retrograde Signaling for Metabolic Control of Aging and Histone Acetylation in Respiratory-Defective Cells", Cell Reports, vol. 7, pp. 565-574, 2014. http://dx.doi.org/10.1016/j.celrep.2014.03.029
- G. Mariño, F. Pietrocola, T. Eisenberg, Y. Kong, S. Malik, A. Andryushkova, S. Schroeder, T. Pendl, A. Harger, M. Niso-Santano, N. Zamzami, M. Scoazec, S. Durand, D. Enot, �. Fernández, I. Martins, O. Kepp, L. Senovilla, C. Bauvy, E. Morselli, E. Vacchelli, M. Bennetzen, C. Magnes, F. Sinner, T. Pieber, C. López-Otín, M. Maiuri, P. Codogno, J. Andersen, J. Hill, F. Madeo, and G. Kroemer, "Regulation of Autophagy by Cytosolic Acetyl-Coenzyme A", Molecular Cell, vol. 53, pp. 710-725, 2014. http://dx.doi.org/10.1016/j.molcel.2014.01.016
- P. Henriksen, S.A. Wagner, B.T. Weinert, S. Sharma, G. Bačinskaja, M. Rehman, A.H. Juffer, T.C. Walther, M. Lisby, and C. Choudhary, "Proteome-wide Analysis of Lysine Acetylation Suggests its Broad Regulatory Scope in Saccharomyces cerevisiae", Molecular & Cellular Proteomics, vol. 11, pp. 1510-1522, 2012. http://dx.doi.org/10.1074/mcp.M112.017251
- W. Kaelin, and S. McKnight, "Influence of Metabolism on Epigenetics and Disease", Cell, vol. 153, pp. 56-69, 2013. http://dx.doi.org/10.1016/j.cell.2013.03.004
- C. Lu, and C. Thompson, "Metabolic Regulation of Epigenetics", Cell Metabolism, vol. 16, pp. 9-17, 2012. http://dx.doi.org/10.1016/j.cmet.2012.06.001
- C.B. Millar, and M. Grunstein, "Genome-wide patterns of histone modifications in yeast", Nature Reviews Molecular Cell Biology, vol. 7, pp. 657-666, 2006. http://dx.doi.org/10.1038/nrm1986
- T. Eisenberg, H. Knauer, A. Schauer, S. Büttner, C. Ruckenstuhl, D. Carmona-Gutierrez, J. Ring, S. Schroeder, C. Magnes, L. Antonacci, H. Fussi, L. Deszcz, R. Hartl, E. Schraml, A. Criollo, E. Megalou, D. Weiskopf, P. Laun, G. Heeren, M. Breitenbach, B. Grubeck-Loebenstein, E. Herker, B. Fahrenkrog, K. Fröhlich, F. Sinner, N. Tavernarakis, N. Minois, G. Kroemer, and F. Madeo, "Induction of autophagy by spermidine promotes longevity", Nature Cell Biology, vol. 11, pp. 1305-1314, 2009. http://dx.doi.org/10.1038/ncb1975
- D. Rubinsztein, G. Mariño, and G. Kroemer, "Autophagy and Aging", Cell, vol. 146, pp. 682-695, 2011. http://dx.doi.org/10.1016/j.cell.2011.07.030
- A.M. Cuervo, "Autophagy and aging: keeping that old broom working", Trends in Genetics, vol. 24, pp. 604-612, 2008. http://dx.doi.org/10.1016/j.tig.2008.10.002
- M.A. van den Berg, P. de Jong-Gubbels, C.J. Kortland, J.P. van Dijken, J.T. Pronk, and H.Y. Steensma, "The Two Acetyl-coenzyme A Synthetases of Saccharomyces cerevisiae Differ with Respect to Kinetic Properties and Transcriptional Regulation", Journal of Biological Chemistry, vol. 271, pp. 28953-28959, 1996. http://dx.doi.org/10.1074/jbc.271.46.28953
- H. Takahashi, J.M. McCaffery, R.A. Irizarry, and J.D. Boeke, "Nucleocytosolic Acetyl-Coenzyme A Synthetase Is Required for Histone Acetylation and Global Transcription", Molecular Cell, vol. 23, pp. 207-217, 2006. http://dx.doi.org/10.1016/j.molcel.2006.05.040
- C.B. Fleck, and M. Brock, "Re-characterisation of Saccharomyces cerevisiae Ach1p: Fungal CoA-transferases are involved in acetic acid detoxification", Fungal Genetics and Biology, vol. 46, pp. 473-485, 2009. http://dx.doi.org/10.1016/j.fgb.2009.03.004
- J.T. Pronk, H. Yde Steensma, and J.P. Van Dijken, "Pyruvate metabolism in Saccharomyces cerevisiae.", Yeast (Chichester, England), 1996. http://www.ncbi.nlm.nih.gov/pubmed/9123965
- D.K. Bricker, E.B. Taylor, J.C. Schell, T. Orsak, A. Boutron, Y. Chen, J.E. Cox, C.M. Cardon, J.G. Van Vranken, N. Dephoure, C. Redin, S. Boudina, S.P. Gygi, M. Brivet, C.S. Thummel, and J. Rutter, "A Mitochondrial Pyruvate Carrier Required for Pyruvate Uptake in Yeast, Drosophila , and Humans", Science, vol. 337, pp. 96-100, 2012. http://dx.doi.org/10.1126/science.1218099
- S. Herzig, E. Raemy, S. Montessuit, J. Veuthey, N. Zamboni, B. Westermann, E.R.S. Kunji, and J. Martinou, "Identification and Functional Expression of the Mitochondrial Pyruvate Carrier", Science, vol. 337, pp. 93-96, 2012. http://dx.doi.org/10.1126/science.1218530
- M. Casal, S. Paiva, O. Queirós, and I. Soares-Silva, "Transport of carboxylic acids in yeasts", FEMS Microbiology Reviews, vol. 32, pp. 974-994, 2008. http://dx.doi.org/10.1111/j.1574-6976.2008.00128.x
- I. Orlandi, R. Ronzulli, N. Casatta, and M. Vai, "Ethanol and Acetate Acting as Carbon/Energy Sources Negatively Affect Yeast Chronological Aging", Oxidative Medicine and Cellular Longevity, vol. 2013, pp. 1-10, 2013. http://dx.doi.org/10.1155/2013/802870
- O. de Smidt, J.C. du Preez, and J. Albertyn, "The alcohol dehydrogenases ofSaccharomyces cerevisiae: a comprehensive review", FEMS Yeast Research, vol. 8, pp. 967-978, 2008. http://dx.doi.org/10.1111/j.1567-1364.2008.00387.x
- F. Saint-Prix, L. Bönquist, and S. Dequin, "Functional analysis of the ALD gene family of Saccharomyces cerevisiae during anaerobic growth on glucose: the NADP+-dependent Ald6p and Ald5p isoforms play a major role in acetate formation", Microbiology, vol. 150, pp. 2209-2220, 2004. http://dx.doi.org/10.1099/mic.0.26999-0
- P. Jong-Gubbels, M.A. Berg, M.A. Luttik, H. Steensma, J.P. Dijken, and J.T. Pronk, "Overproduction of acetyl-coenzyme A synthetase isoenzymes in respiringSaccharomyces cerevisiaecells does not reduce acetate production after exposure to glucose excess", FEMS Microbiology Letters, vol. 165, pp. 15-20, 1998. http://dx.doi.org/10.1111/j.1574-6968.1998.tb13121.x
- F. Remize, E. Andrieu, and S. Dequin, "Engineering of the Pyruvate Dehydrogenase Bypass inSaccharomyces cerevisiae: Role of the Cytosolic Mg2+and Mitochondrial K+Acetaldehyde Dehydrogenases Ald6p and Ald4p in Acetate Formation during Alcoholic Fermentation", Applied and Environmental Microbiology, vol. 66, pp. 3151-3159, 2000. http://dx.doi.org/10.1128/AEM.66.8.3151-3159.2000
- Y.J. Lee, J.W. Jang, K.J. Kim, and P.J. Maeng, "TCA cycle‐independent acetate metabolism via the glyoxylate cycle in Saccharomyces cerevisiae", Yeast, vol. 28, pp. 153-166, 2010. http://dx.doi.org/10.1002/yea.1828
- E. Boles, P. de Jong-Gubbels, and J.T. Pronk, "Identification and characterization of MAE1, the Saccharomyces cerevisiae structural gene encoding mitochondrial malic enzyme.", Journal of bacteriology, 1998. http://www.ncbi.nlm.nih.gov/pubmed/9603875
- M.M. dos Santos, A.K. Gombert, B. Christensen, L. Olsson, and J. Nielsen, "Identification of In Vivo Enzyme Activities in the Cometabolism of Glucose and Acetate by Saccharomyces cerevisiae by Using 13 C-Labeled Substrates", Eukaryotic Cell, vol. 2, pp. 599-608, 2003. http://dx.doi.org/10.1128/EC.2.3.599-608.2003
- L. Palmieri, F.M. Lasorsa, A. De Palma, F. Palmieri, M.J. Runswick, and J.E. Walker, "Identification of the yeast ACR1 gene product as a succinate‐fumarate transporter essential for growth on ethanol or acetate", FEBS Letters, vol. 417, pp. 114-118, 1997. http://dx.doi.org/10.1016/S0014-5793(97)01269-6
- N. Casatta, A. Porro, I. Orlandi, L. Brambilla, and M. Vai, "Lack of Sir2 increases acetate consumption and decreases extracellular pro-aging factors", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1833, pp. 593-601, 2013. http://dx.doi.org/10.1016/j.bbamcr.2012.11.008
- Y. Lin, J. Lu, J. Zhang, W. Walter, W. Dang, J. Wan, S. Tao, J. Qian, Y. Zhao, J.D. Boeke, S.L. Berger, and H. Zhu, "Protein Acetylation Microarray Reveals that NuA4 Controls Key Metabolic Target Regulating Gluconeogenesis", Cell, vol. 136, pp. 1073-1084, 2009. http://dx.doi.org/10.1016/j.cell.2009.01.033
- K. Guan, and Y. Xiong, "Regulation of intermediary metabolism by protein acetylation", Trends in Biochemical Sciences, vol. 36, pp. 108-116, 2011. http://dx.doi.org/10.1016/j.tibs.2010.09.003
- C. Choudhary, B.T. Weinert, Y. Nishida, E. Verdin, and M. Mann, "The growing landscape of lysine acetylation links metabolism and cell signalling", Nature Reviews Molecular Cell Biology, vol. 15, pp. 536-550, 2014. http://dx.doi.org/10.1038/nrm3841
- L. Cai, B. Sutter, B. Li, and B. Tu, "Acetyl-CoA Induces Cell Growth and Proliferation by Promoting the Acetylation of Histones at Growth Genes", Molecular Cell, vol. 42, pp. 426-437, 2011. http://dx.doi.org/10.1016/j.molcel.2011.05.004
- A.J. Koning, P.Y. Lum, J.M. Williams, and R. Wright, "DiOC6 staining reveals organelle structure and dynamics in living yeast cells", Cell Motility, vol. 25, pp. 111-128, 1993. http://dx.doi.org/10.1002/cm.970250202
- D.A. Knorre, K.Y. Popadin, S.S. Sokolov, and F.F. Severin, "Roles of Mitochondrial Dynamics under Stressful and Normal Conditions in Yeast Cells", Oxidative Medicine and Cellular Longevity, vol. 2013, pp. 1-6, 2013. http://dx.doi.org/10.1155/2013/139491
- S. Merz, M. Hammermeister, K. Altmann, M. Dürr, and B. Westermann, "Molecular machinery of mitochondrial dynamics in yeast", bchm, vol. 388, pp. 917-926, 2007. http://dx.doi.org/10.1515/BC.2007.110
- M. Breitenbach, M. Rinnerthaler, J. Hartl, A. Stincone, J. Vowinckel, H. Breitenbach-Koller, and M. Ralser, "Mitochondria in ageing: there is metabolism beyond the ROS", FEMS Yeast Research, vol. 14, pp. 198-212, 2014. http://dx.doi.org/10.1111/1567-1364.12134
- A.B. Demir, and A. Koc, "Assessment of chronological lifespan dependent molecular damages in yeast lacking mitochondrial antioxidant genes", Biochemical and Biophysical Research Communications, vol. 400, pp. 106-110, 2010. http://dx.doi.org/10.1016/j.bbrc.2010.08.019
- C.W. van Roermund, E.H. Hettema, M. van den Berg, H.F. Tabak, and R.J. Wanders, "Molecular characterization of carnitine-dependent transport of acetyl-CoA from peroxisomes to mitochondria in Saccharomyces cerevisiae and identification of a plasma membrane carnitine transporter, Agp2p.", The EMBO journal, 1999. http://www.ncbi.nlm.nih.gov/pubmed/10545096
- J.H. Swiegers, N. Dippenaar, I.S. Pretorius, and F.F. Bauer, "Carnitine‐dependent metabolic activities in Saccharomyces cerevisiae: three carnitine acetyltransferases are essential in a carnitine‐dependent strain", Yeast, vol. 18, pp. 585-595, 2001. http://dx.doi.org/10.1002/yea.712
- L. Palmieri, F.M. Lasorsa, V. Iacobazzi, M.J. Runswick, F. Palmieri, and J.E. Walker, "Identification of the mitochondrial carnitine carrier in Saccharomyces cerevisiae", FEBS Letters, vol. 462, pp. 472-476, 1999. http://dx.doi.org/10.1016/S0014-5793(99)01555-0
- I. Orlandi, N. Casatta, and M. Vai, "Lack of Ach1 CoA-Transferase Triggers Apoptosis and Decreases Chronological Lifespan in Yeast", Frontiers in Oncology, vol. 2, 2012. http://dx.doi.org/10.3389/fonc.2012.00067
- T. Eisenberg, S. Schroeder, S. Büttner, D. Carmona-Gutierrez, T. Pendl, A. Andryushkova, G. Mariño, F. Pietrocola, A. Harger, A. Zimmermann, C. Magnes, F. Sinner, S. Sedej, T.R. Pieber, J. Dengjel, S. Sigrist, G. Kroemer, and F. Madeo, "A histone point mutation that switches on autophagy", Autophagy, vol. 10, pp. 1143-1145, 2014. http://dx.doi.org/10.4161/auto.28767
- K. Suzuki, S. Nakamura, M. Morimoto, K. Fujii, N.N. Noda, F. Inagaki, and Y. Ohsumi, "Proteomic Profiling of Autophagosome Cargo in Saccharomyces cerevisiae", PLoS ONE, vol. 9, pp. e91651, 2014. http://dx.doi.org/10.1371/journal.pone.0091651
- J. Onodera, and Y. Ohsumi, "Ald6p Is a Preferred Target for Autophagy in Yeast, Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 279, pp. 16071-16076, 2004. http://dx.doi.org/10.1074/jbc.M312706200
- P. Fabrizio, C. Gattazzo, L. Battistella, M. Wei, C. Cheng, K. McGrew, and V.D. Longo, "Sir2 Blocks Extreme Life-Span Extension", Cell, vol. 123, pp. 655-667, 2005. http://dx.doi.org/10.1016/j.cell.2005.08.042
- V.M. Boer, S. Amini, and D. Botstein, "Influence of genotype and nutrition on survival and metabolism of starving yeast", Proceedings of the National Academy of Sciences, vol. 105, pp. 6930-6935, 2008. http://dx.doi.org/10.1073/pnas.0802601105
- M. Vanoni, M. Vai, L. Popolo, and L. Alberghina, "Structural heterogeneity in populations of the budding yeast Saccharomyces cerevisiae.", Journal of bacteriology, 1983. http://www.ncbi.nlm.nih.gov/pubmed/6358196
- A.B. Canelas, C. Ras, A. ten Pierick, J.C. van Dam, J.J. Heijnen, and W.M. van Gulik, "Leakage-free rapid quenching technique for yeast metabolomics", Metabolomics, vol. 4, pp. 226-239, 2008. http://dx.doi.org/10.1007/s11306-008-0116-4
- A.B. Canelas, A. ten Pierick, C. Ras, R.M. Seifar, J.C. van Dam, W.M. van Gulik, and J.J. Heijnen, "Quantitative Evaluation of Intracellular Metabolite Extraction Techniques for Yeast Metabolomics", Analytical Chemistry, vol. 81, pp. 7379-7389, 2009. http://dx.doi.org/10.1021/ac900999t
- G. Agrimi, L. Brambilla, G. Frascotti, I. Pisano, D. Porro, M. Vai, and L. Palmieri, "Deletion or Overexpression of Mitochondrial NAD + Carriers in Saccharomyces cerevisiae Alters Cellular NAD and ATP Contents and Affects Mitochondrial Metabolism and the Rate of Glycolysis", Applied and Environmental Microbiology, vol. 77, pp. 2239-2246, 2011. http://dx.doi.org/10.1128/AEM.01703-10
- A. Aranda, and M. del Olmo, "Response to acetaldehyde stress in the yeast Saccharomyces cerevisiae involves a strain‐dependent regulation of several ALD genes and is mediatedby the general stress response pathway", Yeast, vol. 20, pp. 747-759, 2003. http://dx.doi.org/10.1002/yea.991
- E. Postma, C. Verduyn, W.A. Scheffers, and J.P. Van Dijken, "Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiae.", Applied and environmental microbiology, 1989. http://www.ncbi.nlm.nih.gov/pubmed/2566299
- F. Madeo, E. Fröhlich, M. Ligr, M. Grey, S.J. Sigrist, D.H. Wolf, and K. Fröhlich, "Oxygen Stress: A Regulator of Apoptosis in Yeast", The Journal of Cell Biology, vol. 145, pp. 757-767, 1999. http://dx.doi.org/10.1083/jcb.145.4.757
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
The authors are grateful to Neil Campbell for English editing. We acknowledge funding of the project SysBioNet, Italian Roadmap Research Infrastructures.
COPYRIGHT
© 2014

Rewiring yeast acetate metabolism through MPC1 loss of function leads to mitochondrial damage and decreases chronological lifespan by Ivan Orlandi et al. is licensed under a Creative Commons Attribution 4.0 International License.








