
Research Articles:
Microbial Cell, Vol. 3, No. 8, pp. 338 - 351; doi: 10.15698/mic2016.08.518
Attenuation of polyglutamine-induced toxicity by enhancement of mitochondrial OXPHOS in yeast and fly models of aging
1 Neuroscience Graduate Program, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
2 Department of Neurology, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
3 Department of Biochemistry and Molecular Biology, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
4 Department of Molecular and Cellular Pharmacology, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
5 Molecular and Cellular Pharmacology Graduate Program, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
6 Human Genetics and Genomics Graduate Program, University of Miami Miller School of Medicine, Miami, FL 33136, USA.
¶ Current Address: Gene Expression Laboratory. The Salk Institute for Biological Studies, La Jolla, California 92037, USA.
# Current Address: Department of Neurology, School of Medicine, Johns Hopkins University, Maryland 21205, USA.
Keywords: Saccharomyces cerevisiae, mitochondrial respiration, mitochondrial OXPHOS, mitochondrial biogenesis, polyglutamine toxicity, yeast chronological life span, Drosophila model, caloric restriction.
Abbreviations:
CLS - chronological life span,
polyQ – polyglutamine,
OE – overexpression,
OXPHOS - oxidative phosphorylation.
Received originally: 17/05/2016 Received in revised form: 23/06/2016
Accepted: 04/07/2016
Published: 26/07/2016
Correspondence:
Antoni Barrientos, Departments of Neurology and Biochemistry & Molecular Biology, University of Miami Miller School of Medicine; RMSB 2067, 1600 NW 10th Ave., Miami, FL 33136, USA abarrientos@med.miami.edu
Conflict of interest statement: The authors declare no conflict of interest.
Please cite this article as: Andrea L. Ruetenik, Alejandro Ocampo, Kai Ruan, Yi Zhu, Chong Li, R. Grace Zhai and Antoni Barrientos (2016). Attenuation of polyglutamine-induced toxicity by enhancement of mitochondrial OXPHOS in yeast and fly models of aging. Microbial Cell 3(8): 338-351.
Abstract
Defects in mitochondrial biogenesis and function are common in many neurodegenerative disorders, including Huntington’s disease (HD). We have previously shown that in yeast models of HD, enhancement of mitochondrial biogenesis through overexpression of Hap4, the catalytic subunit of the transcriptional complex that regulates mitochondrial gene expression, alleviates the growth arrest induced by expanded polyglutamine (polyQ) tract peptides in rapidly dividing cells. However, the mechanism through which HAP4 overexpression exerts this protection remains unclear. Furthermore, it remains unexplored whether HAP4 overexpression and increased respiratory function during growth can also protect against polyQ-induced toxicity during yeast chronological lifespan. Here, we show that in yeast, mitochondrial respiration and oxidative phosphorylation (OXPHOS) are essential for protection against the polyQ-induced growth defect by HAP4 overexpression. In addition, we show that not only increased HAP4 levels, but also alternative interventions, including calorie restriction, that result in enhanced mitochondrial biogenesis confer protection against polyQ toxicity during stationary phase. The data obtained in yeast models guided experiments in a fly model of HD, where we show that enhancement of mitochondrial biogenesis can also protect against neurodegeneration and behavioral deficits. Our results suggest that therapeutic interventions aiming at the enhancement of mitochondrial respiration and OXPHOS could reduce polyQ toxicity and delay disease onset.
INTRODUCTION
Metabolic and mitochondrial abnormalities are a prominent feature of aging and neurodegeneration, which is not surprising, given the high-energy demands of neuronal function. Mitochondria perform multiple roles essential for cellular metabolism and physiology. Their main function is the conversion of energy stored in nutrients into chemical energy, aerobically, through oxidative phosphorylation (OXPHOS), which couples oxygen reduction by the mitochondrial respiratory chain (MRC) to ATP synthesis.
–
Neurodegenerative diseases are frequently caused by the gain-of-toxic-function of disease-specific proteins that misfold and oligomerize, affecting the integrity and function of selective neuronal systems. Despite toxic protein heterogeneity, mounting evidence suggest that mitochondrial dysfunction and oxidative stress occur early in all major neurodegenerative diseases [1], including α-synucleopathies, taupathies, and polyglutamine (polyQ) disorders. PolyQ disorders, including HD and spinocerebellar ataxias, are caused by a CAG codon repeat expansion in disease-specific genes resulting in the expression of misfolding/aggregation-prone proteins with expanded polyQ stretches. Mitochondrial dysfunction, altered mitochondrial dynamics and impaired axonal trafficking have been associated with the pathogenesis of polyQ diseases in human patients and several research model organisms [2][3]. In addition, mutant huntingtin (Htt) may damage neurons directly by inducing mitochondrial depolarization and altering calcium homeostasis in patients and in mouse models [2]. Finally, mutant Htt has been shown to alter mitochondrial function indirectly by inhibiting expression of the transcriptional co-activator PGC-1α (peroxisome proliferator-activated receptor (PPAR)-gamma-coactivator 1α, which regulates several metabolic processes including mitochondrial biogenesis and respiration when interacting with the respiratory factors NRF1 and NRF2 [3].
–
Due to the complexity of proteotoxic neuronal death, development of suitable models is critical to dissect the precise mechanism/s in the disease process and the aging/disease relationship. Whereas several mammalian cell culture models have been developed over the past 15 years, the unicellular yeast model Saccharomyces cerevisiae and its powerful genetics has also been successfully used to study the protein aggregation associated with polyQ disorders [4][5][6][7]. Although yeast cells lack many of the structural and functional hallmarks of neuronal cells that are critical for the progression of neurodegeneration, yeast models have provided a platform to elucidate the basic cellular mechanisms of toxicity triggered by human neurotoxic proteins and to identify targets for therapeutic intervention [7][8][9]. In S. cerevisiae, expression of Htt exon-1 fragments comprising the polyQ stretches faithfully recapitulates Htt misfolding/aggregation in a polyQ length-dependent manner [10]. Upon polyQ toxicity, several pathways, including ER stress, cytoskeletal disturbances, oxidative stress, and mitochondrial dysfunction contribute to growth arrest and cell death [4][6][11]. Genetic or pharmacological interventions aiming to protect each of these pathways from disruption have been shown to confer protection against polyQ toxicity. Specifically, we have shown that in growing yeast cultures, polyQ-induced toxicity can be suppressed by enhancement of mitochondrial biogenesis, achieved by overexpression of Hap4, the catalytic subunit of the transcriptional complex Hap2,3,4,5 that globally activates transcription of nuclear genes involved in mitochondrial respiration [12]. The specific mechanism involved in the suppression remains to be fully elucidated.
–
As a handicap for the currently available yeast models of proteopathies, all studies reported to date have been conducted on rapidly dividing mitotic cells, and have manifested acute toxicity based on the high galactose-inducible expression of toxic proteins. Only a single study has looked at yeast survival in the stationary phase of yeast expressing α-synuclein, also from a galactose-inducible promoter [13]. Nevertheless, in this report, α-synuclein expression was induced at the moment of inoculation, severely restricting survival during the exponential phase and diauxic shift. While studies in dividing yeast are interesting and have provided useful information into the diverse cellular processes that mutant proteins can alter, these models fail to mirror the post-mitotic state of neurons in the adult brain. Furthermore, due to the fact that neurodegenerative disorders are age-associated disorders, we believe that disease models that facilitate the study of proteotoxicity in the context of aging will be more suitable for the better understanding of these kinds of diseases. These considerations support yeast chronological aging as a good model to study mitochondrial function alterations involved in aging and age-related disorders [8].
–
The yeast chronological life span (CLS) model of aging measures the capacity of stationary (G0) cultures to maintain viability over time (1 to several weeks) [14]. The CLS model of aging is an established model for the regulation of aging in post-mitotic mammalian cells, such as neurons [8]. For CLS studies, yeast cells are usually aged in media containing 2% glucose. Under these conditions, cells divide exponentially producing energy preferentially by fermentation while respiration is repressed in a glucose concentration-dependent manner. As glucose is being consumed, growth slows down and the diauxic shift occurs, which involves a shift from fermentation to respiration. In addition, the activation of stress resistance mechanisms and the accumulation of nutrient stores (glycogen and trehalose) allow the survival in the stationary phase where the metabolic rate is significantly reduced. Mitochondrial respiration during growth is known to be essential for a strain to achieve a standard wild-type CLS [15]. However, yeast cells have a large reserve respiratory capacity to sustain CLS, as respiration only limits CLS when depleted below a threshold of ~40% of wild-type respiration [16]. Strains that respire below this threshold during growth have extremely poor respiratory capacity in the stationary phase and rapidly consume their nutrient stores due to the inefficient production of energy through fermentation, resulting in a very short CLS [16]. How polyQ expression interferes with yeast CLS remains unclear.
–
To study polyQ toxicity in the context of aging and its suppression by enhancement of mitochondrial biogenesis, we have created flexible beta-estradiol inducible yeast models that allow for the tight regulation of toxic protein expression at any moment of growth. Our data show that boosted mitochondrial respiration during growth is essential for Hap4-induced suppression of polyQ toxicity during CLS and that several genetic and nutritional interventions that enhance mitochondrial respiration during growth and extend life span effectively alleviate polyQ toxicity during CLS. These data has been subsequently validated in a Drosophila model of HD, where overexpression of a PGC1-α homolog also protects against neurodegeneration and behavioral deficits. Our results further suggest that therapeutic interventions that enhance mitochondrial OXPHOS could reduce polyQ toxicity and delay disease onset in patients.
RESULTS
Development of yeast models of polyQ cytotoxicity in post-mitotic cells.
In order to study proteotoxicity in post-mitotic yeast using the CLS assay, we generated metabolism-independent inducible models of polyQ toxicity. To this end, we constucted and tested several candidate expression systems [17], and ultimately chose an optimized β-estradiol-inducible system modified from [18] as the system that performed best in our conditions (Figure 1A). This expression system is based on the constitutive expression of a transactivator fusion protein GAL4.ER.VP16, which is formed by a GAL4 DNA binding domain, a β-estradiol receptor domain and a VP16 (virus protein 16) transcriptional activator that can activate transcription of a gene placed under the control of a galactose-inducible promoter (GAL1pr). In the absence of the hormone, the fusion protein is repressed by the yeast chaperones from the Hsp90 family [18]. Upon media supplementation with β-estradiol, the fusion protein is de-repressed, binds to GAL1pr through the GAL4 DNA binding domain and the VP16 recruits the transcriptional machinery to start transcription of the gene of interest.
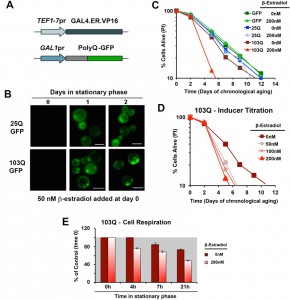 | FIGURE 1: Generation of inducible yeast models of proteopathies. (A) Schematic of polyQ-GFP expression cassettes under the control of the β-estradiol inducible promoter. (B) Chronology of polyQ-GFP protein accumulation, followed by fluorescence microscopy, in cells induced with 50 nM β-estradiol. The bar is 5 µm. (C) and (D) Yeast CLS. Survival of wild-type cells expressing 25Q or 103Q from a β-estradiol-inducible promoter activated with the indicated amounts of inducer or supplemented with the solvent (ethanol) was estimated by propidium iodide (PI) staining and flow cytometry analysis of 10,000 cells. Data is an average of three samples in % of cells alive at day 0 (72 h after inoculation). In (D) a β-estradiol titration was performed. (E) Time-course of endogenous cell respiration during the first day in stationary phase of growth of non-induced and induced 103Q cultures. |
Using this system, we created models expressing 25Q, 46Q, 72Q or 103Q N-terminal Huntingtin (Htt) exon-1 fragment proteins, and have confirmed that polyQ expression is well-induced with a low β-estradiol concentration (50 nM) upon transition to the stationary phase (see 25Q and 103Q in Figure 1B) without detectable leakage of the system before induction (Figure 1B and [17]). For the chronological life span (CLS) experiments described herein, wild-type cells expressing 25Q- and cells expressing highly-pathogenic 103Q-N-terminal Htt, under the control of the described β-estradiol inducible promoter, were pre-grown in minimum media containing 2% glucose until they reached stationary phase (72 hours). At this moment, protein expression was induced by addition of β-estradiol. After induction, polyQ protein accumulates in a diffused distribution in the cytoplasm of 25Q-expressing cells, while 103Q fragments form insoluble protein aggregates over time (Figure 1B). When expression was induced with 200 nM β-estradiol, expression of GFP or 25Q did not produce any measurable effect in CLS, as determined by flow cytometry after PI staining, whereas 103Q produced a toxic effect that curtails maximum yeast CLS by 50%, (Figure 1C). These results were in agreement with the analysis of cellular viability using a clonogenic assay based on counting of CFU (colony formation units, Figure S1). A titration of the inducer indicated that polyQ expression is well induced and only slightly increases further in a dose-dependent manner with concentrations from 50 nM to 200 nM, although 103Q toxic effect on CLS was similar in this concentration range (Figure 1D). As a control, no toxic effects of the hormone were observed (not shown).
–
PolyQ cytotoxicity in chronologically aging yeast is suppressed by genetically-driven enhancement of mitochondrial biogenesis
We have previously reported that cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS function, loss of membrane potential and ROS generation [4][5]. Here, we have observed that 103Q expression induced at day 0 of chronological life span resulted in 25% lowered endogeneous cell respiration after 4 h in stationary phase, when all the cells are still alive (Figure 1E). Although respiration of non-induced cells also decreased over time, induced cells were decreased by 35% after 20 h in stationary phase (Figure 1E and 2A). As a toxicity suppressor mechanism, we have shown earlier that enhancement of mitochondrial biogenesis through overexpression (OE) of Hap4, the catalytic subunit of the HAP transcriptional complex that serves as the master regulator of nuclear genes encoded mitochondrial proteins in yeast [12], confers robust protection against the 103Q-induced growth deficit during the exponential phase [4]. Therefore, we hypothesized that HAP4 may also confer protection against 103Q-induced toxicity in the stationary phase.
–
To test this hypothesis, 103Q cells and 103Q cells carrying a construct that overexpresses HAP4 under the control of its own promoter were grown in media containing 2% glucose and 103Q expression was induced upon transition to the stationary phase. As previously reported [16], non-induced wild-type-like cells constitutively overexpressing HAP4 respired at a rate of 150% of wild-type during growth and 50% of wild-type during the stationary phase (Figure 2A). For clarification, wild-type strains have a reduced metabolic rate when they reach the stationary phase, and their respiratory rate at day 0 of CLS (72 hr after inoculation) – here considered as 100% – is actually 20% of their exponential-phase respiratory rate [16].
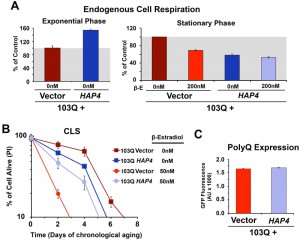 | FIGURE 2: PolyQ-induced toxicity in CLS models of yeast aging is suppressed by HAP4 overexpression-mediated increase in mitochondrial biogenesis. (A) Endogenous cell respiration during the exponential and stationary phases of growth of non-induced and induced 103Q cultures overexpressing or not HAP4. Bars represent mean ± S.D., n = 3. (B) Effect of increased mitochondrial biogenesis by HAP4 overexpression on CLS of yeast expressing 103Q from day 0 in the stationary phase. Error bars represent S.E.M. for 3 independent experiments. (C) PolyQ expression levels at day 1 after β-estradiol induction estimated by flow cytometry quantification of GFP fluorescence. |
HAP4 OE was previosuly reported to slightly extend CLS when the cells were transferred to water upon reaching the stationary phase [19]. However, under our experimental conditions, enhanced mitochondrial biogenesis and respiration during growth was not sufficient to extend maximum CLS in non-induced cells [16] (Figure 2B). On the contrary, enhanced mitochondrial biogenesis was able to prevent the decline in respiratory capacity of 103Q-expressing cells after 20 hours in exponential phase (Figure 2A) and, most importantly, provide a CLS extension to yeast expressing 103Q, from 3 to 5 days (Figure 2B). This suppression of 103Q toxicity was not due to decreased 103Q protein, as polyQ-GFP fluorescence was indistinguishable between cells with or without HAP4 expression (Figure 2C). We envision that by increasing mitochondrial biogenesis during growth, the cell becomes equipped with a larger buffering system against 103Q-induced mitochondrial damage later during CLS [4]. Furthermore, preserving aerobic energy conversion is expected to have a positive impact in ATP-dependent 103Q clearance pathways and refolding chaperone systems.
–
PolyQ cytotoxicity in chronologically aging yeast is attenuated by nutritional respiratory preconditioning
Yeast CLS has been reported to be extended by increasing the levels of cellular respiration during the initial exponential growth phase achieved by culture on a respiratory carbon source [4][16][19][20][21][22] or by pre-culturing the cells in media containing 0.5% rather than 2% glucose (caloric restriction or CR) [16][23][24][25]. Although the mechanisms involved in the two models are very different, as explained below, we wanted to test whether suppression of 103Q toxicity during CLS could also be confered by these interventions known to enhance mitochondrial respiration and biogenesis.
–
First, we started by modifying the synthetic medium from containing glucose (WOGLU), a fermentable carbon source, to containing the non-fermentable carbon sources ethanol and glycerol (WOEG). In the presence of glucose, mitochondrial biogenesis is repressed in a concentration-dependedt manner through inhibition of the HAP complex [26]. On the contrary, growth in WOEG media results in HAP-mediated increase in mitochondrial biogenesis, including components of the mitochondrial respiratory chain and oxidative phosphorylation system [27][28], similar to what is achieved by overexpression of HAP4. This was reflected by a 2-fold higher endogenous cell respiration during growth in WOEG than in WOGLU (Figure 3A). Similarily to the results obtained with HAP4 OE, growth in WOEG media suppressed 103Q toxicity by preventing the decline in respiratory capacity of 103Q-expressing cells after 20 h in exponential phase (Figure 3A) leading to a 2.3-fold increase in maximum survival of 103Q-expressing cells (Figure 3B). This protective effect was not due to a reduction in the levels of polyQ protein that were actually higher in WOEG media compared to WOGLU, as estimated by GFP fluroescence (Figure 3C). The CLS extension conferred by growth in respiratory carbon sources has been suggested to be a reflection of efficient survival in stationary phase requiring respiratory metabolism [29] and a reduction in medium acidification [21]. Furthermore, respiration-adapted cultures, do not just display maximal longevity, but also maintain full replicative lifespan during CLS [19]. Regarding 103Q-expressing cells, we envision, as for HAP4 OE cultures, that enhancement of mitochondrial biogenesis and respiration, could minimize the effect of 103Q-induced mitochondrial damage and might enhance the activity of 103Q refolding chaperone systems.
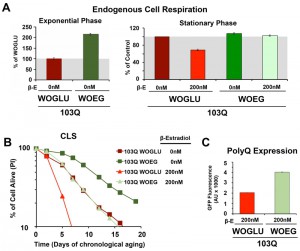 | FIGURE 3: PolyQ-induced toxicity in CLS models of yeast aging is minimized by pre-conditioning the cells in respiratory media. (A) Endogenous cell respiration during the exponential and stationary phases of growth of non-induced and induced 103Q cultures grown in minimum synthetic media containing either the fermentable glucose (WOGLU) or the non-fermentable ethanol-glycerol (WOEG) as the carbon sources. Error bars are S.D., n = 3. *P < 0.05, **P < 0.005. (B) Effect of growth in WOEG respiratory media on 103Q yeast CLS compared to WOGLU media. S.D <1 for all samples, n = 3. |
Subsequently, we decided to test whether caloric restriction (CR) could also suppress 103Q toxicity. In yeast, CR, modeled by growing cells in media with 0.5% glucose, has been shown to result in a Hap4-dependent enhancement of mitochondrial biogenesis, as well as an increase in respiration (Figure 4A and [16]) and ROS-adaptive signaling during growth [16]. CR extends CLS in part through the downregulation of signaling pathways involving nutrient-responsive kinases, such as Ras/cAMP/PKA, TOR, and Sch9, which promote the expression of stress-response genes and the accumulation of storage carbohydrates [16][23][25]. We recently reported that CR-induced extension of CLS required the cells to have a minimum threshold respiratory capacity (40% of wild-type) during exponential growth in non-restricted WOGLU media [16]. In wild-type cells [16] or in our non-induced 103Q cells (Figure 4A), CR led to a 30% higher respiratory rate during exponential growth but, importantly, completely abolished the need for respiration in the stationary phase. CR involves a metabolic remodeling that includes the shift from fermentation to respiration, which is essential for CLS extension [24]. However, the metabolic remodeling that occurs during the diauxic shift allows CR cells to dramatically reduce their metabolic rate in the stationary phase and hence consume their stored nutrients at a rate slower than that of cells grown in 2% glucose [16][30]. Furthermore, in stationary phase, CR cells have lowered oxidative stress [16][20], which may contribute to CLS extension.
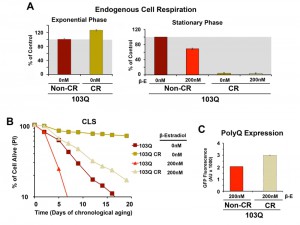 | FIGURE 4: PolyQ-induced toxicity in CLS models of yeast aging is markedly suppressed by pre-conditioning the cells in 0.5% glucose-containing media (caloric restriction or CR). (A) Endogenous cell respiration during the exponential and stationary phases of growth of non-induced and induced 103Q cultures grown in minimum synthetic (WOGLU) media containing either 2% (control) or 0.5% (CR) glucose as the carbon source. Bars represent mean ± S.D., n = 3. (B) Effect of CR in 103Q yeast CLS. S.D <1 for all samples, n = 3. (C) PolyQ expression levels at day 1 after β-estradiol induction estimated by flow cytometry quantification of GFP fluorescence. |
As expected, CR-treated non-induced cells had a markedly longer lifespan than those grown without the intervention (Figure 4B), which is in agreement with previous reports showing that, in wild-type cells, CR led to a more than 2-fold extension of their maximum CLS to 27 days [16][23]. Induction of 103Q-expression at day 0 of CLS in CR-treated cells was still toxic and reduced maximum CLS to slightly over 20 days. However, attention should be focused on the fact that 103Q-expressing cells grown in CR media increased cell survival from 7 days to more than 20 days, an extenstion over that of even non-induced cells growing in non-CR medium (Figure 4B).
–
Because mitochondrial respiration is not required for CLS in CR-treated cells, our data indicate that 103Q toxicity during CLS involves multiorganelle pathways as it does in growing cells. As for the anti-toxicity interventions discussed in previous paragraphs, the CR-treatment effect was not due to decreases in polyQ protein, since, like for cells grown in WOEG, polyQ expression was estimated to be significantly higher in CR cells than those grown in standard glucose-containing media (Figure 4C).
–
Mitochondrial respiration is essential for suppression of PolyQ toxicity via enhanced mitochondrial biogenesis
Although all the anti-103Q toxicity interventions presented in the previous sections involve increases in cellular respiration during growth, we wanted to further explore whether the mitochondrial contribution to this protection involves OXPHOS or if it may relate to some other pathway affected by enhancement of mitochondrial biogenesis. In order to test whether mitochondrial respiration is required for HAP4-mediated suppression of 103Q proteotoxicity, we turned to our mitotic models of galactose-induced polyQ expression during exponential growth. For these assays we used strains expressing either non-toxic 25Q or toxic 103Q motifs further modified to overexpress HAP4 or to carry an empty episomal plasmid (EP). Here, we abolished mitochondrial OXPHOS function by genetic and pharmacologic manipulations and determined the efficacy of HAP4-mediated suppression of 103Q-induced cytotoxicities.
–
First, we used previously reported 103Q yeast, with or without the overexpression of HAP4, that had been completely depleted of mitochondrial DNA (mtDNA) and therefore are rho0 strains [31]. The mtDNA encodes essential components of the OXPHOS system, and, therefore, rho0 yeast are unable to grow in complete media in the presence of respiratory substrates such as ethanol and glycerol (YPEG). Because rho0 cells do not grow efficiently in galactose-containing media, we used the non-repressible raffinose as the fermentable carbon source and induced protein expression when required by supplementing the plates with 0.25% galactose. As shown in Figure 5, HAP4 OE suppressed 103Q-induced toxicity, as previously reported [4]. As expected, the suppression was only effective in rho+ strains, containing mtDNA, but not in rho0 strains, suggesting that HAP4-mediated suppression requires functional mitochondrial respiration.
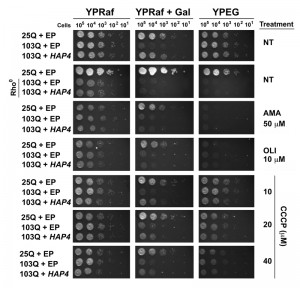 | FIGURE 5: Mitochondrial respiration is essential for suppression of polyQ-induced toxicity by HAP4 overexpression. Serial dilutions of yeast strains on non-inducing media (YPRaf), polyQ-inducing media (YPRaf+Gal), and respiratory media (YPEG) after 2 days of growth at 30°C. Rho0 indicates cells without mitochondrial DNA. EP, Empty plasmid; NT, no treatment (NT); AMA, antimycin A; CCCP, Carbonyl cyanide m-chlorophenyl hydrazone; OLI, oligomycin. |
In addition to the genetic approach, we also pharmacologically inhibited mitochondrial respiratory complex III activity with antimycin A (AMA) that prevents growth in respiratory media (YPEG). AMA treatment abolished the suppression by HAP4 OE (Figure 5). Similar outcomes were obtained when mitochondrial OXPHOS was impaired by supplementation of the growth media with oligomycin (OLI), an inhibitor of the F1Fo ATPase. We next treated the cells with increasing concentrations of the ionophore carbonyl cyanide m-chlorophenyl hydrazine (CCCP). CCCP causes an uncoupling of the mitochondrial proton gradient from ATP production, causing a futile increase in mitochondrial respiration. Although still effective, a reduction in the protective effects of HAP4 OE in 103Q-expressing cells was seen with increasing concentrations of CCCP (Figure 5). These results strongly indicate that coupled mitochondrial respiration is the essential component of the HAP4 mechanism of protection against proteotoxicity in yeast.
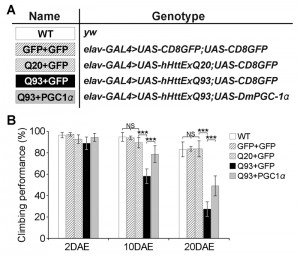 | FIGURE 6: DmPGC-1α/spargel protects against hHttExQ93-induced climbing defects. (A) List of genotypes of the transgenic flies examined. The first exon of human huntingtin gene with 20 (UAS-hHttExQ20) or 93 (UAS-hHttExQ93) repeats of polyQ, and GFP (UAS-CD8GFP) or DmPGC-1α/spargel (UAS-DmPGC-1α) were expressed by the pan-neuronal driver elavC155-GAL4. (B) Climbing performance of wild-type, GFP over-expressing flies, and hHtt-expressing flies that co-expressed GFP or PCG-1α at the ages of 2, 10 and 20 DAE (days after eclosion). Ten groups (10 flies in each, total one hundred flies) of each genotype and age were tested. All data were presented as mean ± S.D. n = 10. Significance level was established by One-Way ANOVA post hoc Tukey’s test. *** P < 0.001. |
Attenuation of PolyQ-induced neurodegeneration in a fly model of Huntington’s disease via enhanced mitochondrial biogenesis
To test whether our results in yeast could be replicated in higher organisms, we turned to a Drosophila model of HD. Similar to the function of the Hap2/3/4/5 complex in yeast, PGC-1α in higher organisms is a transcriptional activator critical in regulating mitochondrial biogenesis [25]. In Drosophila, overexpression of the PGC-1α homolog (DmPGC-1/spargel) has been shown to be sufficient to increase mitochondrial OXPHOS activity, which leads to an extension of fly life span and improvement in tissue homeostasis in aged flies [32]. Here, we used a pan-neuronal driver elavC155-GAL4 to express the exon 1 of human huntingtin gene with pathological 93 polyQ repeats (hHttExQ93) or non-pathological 20 polyQ repeats (hHttExQ20) in the fly nervous system [33]. First, we tested the locomotor activity of the flies by a negative geotaxis assay. As shown in Figure 6, while flies expressing hHttExQ20 showed similar climbing behavior as wild-type flies (yw), expression of hHttExQ93 caused an age-dependent decline of the climbing performance that was significant at 10 and 20 days after eclosion (DAE). This decline of climbing performance was suppressed by co-expression of DmPGC-1α. Consistent with a previous report [32], overexpression of DmPGC-1α enhanced mitochondrial biogenesis by 50% in fly brains, as monitored by the steady state levels of ATP5α, a subunit of mitochondrial F1F0-ATP synthase [34][35] (Figure 7A-B).
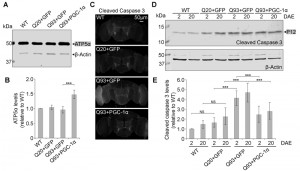 | FIGURE 7: DmPGC-1α/spargel suppresses hHttExQ93-induced activation of cell apoptosis. (A-B) Western blot analysis (A) and quantification (B) of proteins extracted from 2 DAE wild-type, hHttExQ20- or hHttExQ93-overexpressing fly heads. ATP5α was used as a mitochondrial marker. β-Actin was used as an internal control. For quantification, fold change relative to the level of WT group is displayed. (C) Adult (2 DAE) female brains of wild-type, hHttExQ20- or hHttExQ93-overexpressing flies were probed for cleaved caspase-3. Scale bar is 50 µm. (D-E) Western blot analysis (D) and quantification (E) of proteins extracted from 2 DAE and 20 DAE wild-type, hHttExQ20- or hHttExQ93-overexpressing fly heads. For quantification, P12 was considered as cleaved caspase-3 (grey box): -fold change relative to each level in 2 DAE wild-type flies. β-Actin was used as an internal control. All data were presented as mean ± S.D., n = 3. Significance level was established by One-Way ANOVA post hoc Bonferroni test. *** P < 0.001. |
To examine the cellular effect of enhancing mitochondria biogenesis on mutant Htt-induced neurodegeneration, we determined the level of caspase-3 activation, a hallmark of apoptosis [36], at early (2 DAE) and late (20 DAE) ages. Neuronal expression of hHttExQ93 induced an increased level of cleaved caspase-3 throughout the entire fly brain at 2 DAE, which could be suppressed by co-expression of DmPGC-1α (Figure 7C). Therefore, neuronal apoptosis occurred early following hHttExQ93 expression, before the onset of significant decline in climbing behavior. To further quantitatively monitor the activation of apoptosis, we measured the expression level of caspase 3 P12, the activated and fully processed product of pro-caspase 3 [37]. Consistently, expression of hHttExQ93 significantly increased P12 level in the fly brains, which could be attenuated by overexpression of DmPGC-1α at both 2 DAE and 20 DAE (Figure 7D-E).
–
Collectively, these results indicate that enhancing mitochondrial biogenesis by overexpressing DmPGC-1α protects against polyQ-induced neurotoxicity, and this neuroprotective mechanism is likely conserved between yeast and flies.
DISCUSSION
The role of mitochondrial respiration and oxidative phosphorylation in proteotoxic neurodegeneration is a longstanding question, and the mechanism by which enhancement of mitochondrial biogenesis protects against proteotoxicity is still poorly understood. Polyglutamine toxicity is pleiotropic, affecting multiple pathways and organelles, including mitochondria. Among the mitochondrial pathways, our studies using yeast CLS and fly models of neurodegenerative proteopathies identify OXPHOS as the pathway that is most affected by polyglutamine expression and oligomerization. Notably, we reveal that genetic and nutritional interventions that enhance mitochondrial biognesis to precondition cells and tissues with high respiration serve to attenuate polyQ-induced proteotoxicity.
–
Most previous studies of proteotoxicity suppression in yeast models have used rapidly dividing mitotic cells, and have established acute toxicity with a high expression of toxic proteins using galactose-inducible promoters [4][5][6][7][38]. Here, using a β-estadiol expression system capable of inducing toxic polyQ expression at the point of transition to stationary phase [17], we have documented an early mitochondrial respiratory defect. We have also shown that enhancement of mitochondrial biogenesis by overexpression of HAP4 partially suppresses polyQ-induced toxicity in the stationary phase of the CLS assay, a model for neuronal aging, as previosuly reported for the polyQ-induced growth deficit in the exponential phase [4]. Importantly, we report that, in the exponential growth model, anti-polyQ protection by HAP4 overexpression is abolished with the inhibiton of mitochondrial respiration, even though the increase in mitochondrial biogenesis remains, thus demonstrating that protecting OXPHOS pathway is a key for polyQ toxicity attenuation. We have further exploited the yeast paradigm to demonstrate that growth in non-fermentable media and CR media, nutritional interventions known to increase mitochondrial biogenesis and mitochondrial respiration during growth, also confer protection against stationary-phase polyQ toxicity. In the case of CR, multiple additional pathways could be relevant. For example, despite the consensus that mutant polyQ- and alpha-synuclein-induced toxicities involve several distinct pathways, it has been recently reported that CR reduces alpha-synuclein toxicity in aged yeast cells by controlling the maintenance of autophagic activity at homeostatic levels [39]. The possibility exist, however, that the control of autophagy in these growth conditions is a consequence of the CR-induced enhanced OXPHOS function.
–
Over the several experimental settings presented here, we have observed that enhanced respiration during growth results in lowered endogenous cell respiration during stationary phase, the most extreme case being CR-treated cells, which survive without utilizing respiratory metabolism [16]. We have previously shown that higher respiration during growth is associated with higher ROS generation, which can act as signaling molecules to induce an adaptive response in stationary phase involving metabolic remodeling and slow metabolism. This is particularly true for cells grown under CR conditions or in the presence of rapamycin to inhibit the TOR pathway [16][40], and it is also true for cells overexpressing HAP4 [16].
–
We need to emphasize, however, that we performed our respiratory assays using cells in their culture media, and therefore we are measuring “real time” respiration. CR-treated cells maintain a high mitochondrial mass in stationary phase, but this capacity is not utilized for respiration during CLS. The relationship between high respiration during growth phases and chronological life span is also intriguing. An important example are the puf3 mutant strains, which include an increased OXPHOS abundance and respiration during all phases of growth, but a wild-type CLS [41]. In agreement with this, we have shown that increased respiration during growth (e.g., by HAP4 OE) is not sufficient to extend CLS of wild-type cells if it is not accompanied by an enhancement of cell protection systems that promote survival in the stationary phase as for a tor1 mutant- and CR-treated cells [16]. However, in chronologically aged 103Q-expressing cells, in which mitochondrial function is altered, even HAP4 overexpression is able to provide the enhanced mitochondrial mass buffer needed to overcome the polyQ-induced mitochondrial toxicity.
–
As an important validation of the observations made in the yeast models, we have confirmed our HAP4 overexpression results in a fly model of HD, in which we have shown that overexpression of the Drosophila PGC-1α homolog (DmPGC-1/spargel) is also able to protect against the behavioral deficits and neurodegeneration induced by polyQ proteins. It is important to note the potential difference in hHttExQ93 expression levels between our study and previous studies using the same transgenic line [42]. In our study, to ensure similar expression level of hHttExQ93 among all groups, we used UAS-GFP as a control transgenic element. Therefore, the level of hHttExQ93 expression in our study is likely lower than that in previous studies. As a result, the progression of degeneration is slower as hHttExQ93-expressing flies in our study all survived beyond 20 DAE, while a median lifespan of 10 DAE (maximum 17 DAE) was reported in the previous study [42]. The slightly slower degeneration gave us the temporal resolution to assess the age-dependent decline in climbing behavior and the suppression by PGC-1α. Interestingly, we observed the activation of apoptosis before the onset of behavior defects, suggesting apoptosis as an early event and a likely cause of degeneration. Importantly, PGC-1α expression suppresses apoptosis from the early age, providing the molecular basis for the neuroprotection offered by PGC-1α.
–
These results are in agreement with previous studies showing that transcriptional repression of PGC-1α by mutant Htt leads to mitochondrial dysfunction and neurodegeneration in HD mouse models and that exogenous expression of PGC-1α in this model protects against mutant Htt-induced neurotoxicity [3][43]. Furthermore, PGC-1α has been proposed to be a modifier of onset age in HD patients, although functional studies are needed to confirm this and to identify the genetic variations in PGC-1α that enhance or alleviate HD pathogenesis [44][45][46].
–
In conclusion, our results in post-mitotic yeast, using the CLS assay, as well as our results in the Drosophila model of HD, shed new light on how mitochondrial respiration and OXPHOS-dependent pathways are affected by polyQ-induced toxicity, and support the need for further research into the possibile therapeutic potential of interventions aimed at increasing mitochondrial biogenesis in patients suffering from HD and other neurodegenerative proteopathies.
MATERIALS AND METHODS
Yeast strains and methods
Yeast strain and media
The yeast strains used in this study were constructed from the previously reported 25Q and 103Q isogeneic strains, and the respective HAP4 overexpressing strains, with the S. cerevisiae W303-1A background [4][5], modified with the β-estradiol-inducible TEF1-7 promoter described in [17]. Compositions of the different growth media have been described [47][48][49]. Prior to induction with galactose or the indicated amount of β-estradiol, all strains were grown in non-inducible media. For the chronological lifespan assay, strains were grown in the indicated media, using the methods described in [50].
–
Yeast chronological life span determinations
Most chronological life span determinations were performed in cells grown in liquid synthetic complete media containing 2% glucose (WOGLU) supplemented with standard amounts of amino acids and nucleotide bases as previously described including a four fold excess of the supplements tryptophan, leucine, histidine, methionine, adenine and uracyl to avoid possible artifacts due to the auxotrophic deficiencies of the strains [50]. For growth in respiratory media, glucose was substituted by EG (2% ethanol and 2% glycerol). For calorie restriction (CR) experiments, glucose concentration was reduced to 0.5%. Briefly, yeast strains from frozen stock (-80°C) were patched onto complete YPD (2% glucose) or YPEG (2% ethanol and 2% glycerol) agar plates and incubated at 30°C. The following day, cells were inoculated into 10 ml of minimum WO media with the appropriate carbon sources and grown overnight. After 24 hours, cells were inoculated into 50 ml of WO media in 250-ml flasks to an optical density at 600 nm (OD600) of 0.250. Cultures were grown with shaking (250 rpm) at 30°C. Maximum cell density is normally reached after 72 hours of growth in WO, therefore 3 days after inoculation was considered as “day 0” of chronological life span. Subsequently, cellular viability was determined at the indicated days by either propidium iodide (PI) staining followed by flow cytometry analysis or by the colony formation unit (CFU) assay.
–
Propidium iodide staining and flow cytometry analysis
Membrane integrity as a marker of cell viability was assessed by PI staining (Molecular Probes, Eugene, OR, USA) and analyzed by flow cytometry as previously described [50]. Every other day, a sample of stationary phase cultures containing 106 yeast cells per ml in phosphate-buffered saline (PBS) was incubated for 30 min at 30 °C in the presence of 2 μM PI. Flow cytometry analysis was performed on a Becton Dickinson (BD) LSRFortessa™ cell analyzer as described [50]. Excitation was performed using a yellow/green laser at 561 nm; emission was detected using a 20 nm bandpass filter centered at 660 nm (Becton Dickinson, NJ, USA). For each yeast population, three samples of ten thousand cells were analyzed.
–
Colony formation unit assay
To perform the colony formation unit (CFU) assay, cell number was estimated by optical density (OD) for each population and serial dilutions of different cultures were plated onto 3 or 4 YPD plates at an approximate concentration of 100 cells per plate. Plates were incubated at 30°C for 48 h and CFU were counted.
–
Fluorescence microscopy
Wide-field fluorescence microscopy for verification of polyQ-GFP expression after β-estradiol induction in yeast was performed as described [4]. Briefly, we used an Olympus fluorescence BX61 microscope equipped with Nomarski differential interference contrast (DIC) optics, a Uplan Apo 100× objective (NA 1.35), a Roper CoolSnap HQ camera, and Sutter Lambda10-2 excitation and emission filter wheels, and a 175 W Xenon remote source with liquid light guide. Briefly, PI-stained cells were mounted on to slides and examined using a CY3 filter. Images were acquired using SlideBook 4.01 (Intelligent Imaging Innovations, Denver, CO, USA).
–
Cell respiration
Assessment of yeast endogenous cellular respiration was performed polarographically using a Clark-type oxygen electrode (Hansatech Instruments, Norfolk, UK) at 30°C as described [51]. Measurement of respiration in the exponential phase of growth was performed 16 h after concomitant strain inoculation and induction. For exponential phase respiration, cells were grown overnight in the indicated media, reinoculated in fresh media at the same confluence (OD600 = 0.250) and grown for 3-6 h, as indicated, to measure maximal cellular respiration. For stationary phase respiration, cell were treated as describe above and grown for 72 h (day 0) before measure respiration, to allow cell to reach stationary phase. The specific activities reported were corrected for KCN-insensitive respiration.
–
Chemical treatments
Serial dilution growth tests were performed in solid media containing glucose, raffinose or galactose as the carbon source, supplemented or not with several mitochondrial poisons. Plates were supplemented with either 50 μM antimycin A (Sigma) to inhibit the mitochondrial respiratory chain complex III or 10 μM oligomycin (Sigma) to inhibit the mitochondrial F1F0 ATP synthase. Plates were also supplemented with increasing concentrations (10-40 μM) of the uncoupler CCCP (carbonyl cyanide m-chlorophenyl hydrazine). In the “untreated” plates, an equal volume of drug vehicle (ethanol) was added as a control.
–
Drosophila strains and methods
Drosophila strains and culture conditions
All fly strains were maintained on a cornmeal–molasses–yeast medium at room temperature (~22°C) with 60–65% humidity. The following Drosophila strains were used in the studies: elavC155-GAL4 and UAS-CD8GFP were obtained from Bloomington Drosophila Stock Center. UAS-hHttExQ20 and UAS-hHttExQ93 were obtained from Dr. Leslie Thompson [33]. UAS-DmPGC-1α (UAS-srl) was obtained from Dr. Christian Frei [52].
–
Negative geotaxis behavior assay
Ten age-matched female flies from each genotype were placed in a vial marked with a circle 8 cm above the bottom surface. The flies were gently tapped to the bottom and given 10 s to climb. After 10 s, the number of flies that successfully climbed above the 8 cm mark was recorded and divided by the total number of flies. The assay was repeated 10 times and the data were represented as the averaged percentages. Ten independent groups (total 100 flies) per genotype was tested [53].
–
Protein extraction and western blot analysis
Proteins were extracted from fly heads using the homogenizing buffer containing 10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 1 mM dithiothreitol (DTT) and 0.5 mM phenylmethylsulphonyl fluoride (PMSF). Samples were then spun at 14,000g for 30 min at 4°C and the supernatant was collected as supernatant 1. The pellets were resuspended with the protein extraction buffer containing 30 mM HEPES pH 7.9, 0.6 M NaCl, 1.5 mM MgCl2, 0.4 mM EDTA, 1 mM DTT, 25% glycerol, 1% NP-40 and 0.5 mM PMSF and incubated at 4°C for 30 min with vortexing every 6 min. Then the supernatant 2 was collected after 30 min centrifugation at 14,000g at 4°C. Next, supernatants 1 and 2 were equally mixed and heated at 95°C with 4 × Laemmli sample buffer [54]. Samples were loaded onto 15% SDS-polyacrylamide gels and transferred to nitrocellulose membranes. Blots were probed with anti-ATP5α antibody (1:10,000, Abcam), anti-cleaved caspase 3 (1:1,000, Cell Signaling), or anti-β-Actin (1:10,000, Sigma) primary antibodies, and then probed with infrared dye-conjugated IR700 and IR800 secondary antibodies (1:10,000, LI-COR Biosciences). Blots were imaged and processed on an Odyssey Infrared Imaging system.
–
Immunohistochemistry of fly brains
Adult brains were dissected in ice cold PBS (pH 7.4), fixed in 4% formaldehyde for 15 min and washed in PBS with 0.4% Triton X-100 (PBTX). Brains were incubated at 4°C overnight with the following antibodies diluted in PBTX with 5% normal goat serum: anti-cleaved caspase 3 antibody (Asp175; 1:250, Cell Signaling); secondary antibody conjugated to Alexa 488 (1:250, Jackson ImmunoResearch, Molecular Probes). Tissues were then mounted on microscope slides in Vectashield Mounting Medium for Fluorescence (Vector Laboratories).
–
Confocal image acquisition and processing
Confocal microscopy was performed with an Olympus IX81 confocal microscope using an Olympus PlanApo N 20x objective. Images from different genotypes were captured using the same scan setting and parameters. Images were processed using FluoView 10-ASW (Olympus) and assembled using Adobe Photoshop CS6 (Adobe Systems).
–
Statistical Analysis
All experimental measurements were done at least in triplicate. All results are presented as means ± S.E.M. or S.D. as appropriate. Significance testing was performed using the Student’s T-test or One-Way ANOVA, as noted in the figure legends.
References
- M.T. Lin, and M.F. Beal, "Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases", Nature, vol. 443, pp. 787-795, 2006. http://dx.doi.org/10.1038/nature05292
- A.V. Panov, C. Gutekunst, B.R. Leavitt, M.R. Hayden, J.R. Burke, W.J. Strittmatter, and J.T. Greenamyre, "Early mitochondrial calcium defects in Huntington's disease are a direct effect of polyglutamines", Nature Neuroscience, vol. 5, pp. 731-736, 2002. http://dx.doi.org/10.1038/nn884
- L. Cui, H. Jeong, F. Borovecki, C.N. Parkhurst, N. Tanese, and D. Krainc, "Transcriptional Repression of PGC-1α by Mutant Huntingtin Leads to Mitochondrial Dysfunction and Neurodegeneration", Cell, vol. 127, pp. 59-69, 2006. http://dx.doi.org/10.1016/j.cell.2006.09.015
- A. Ocampo, A. Zambrano, and A. Barrientos, "Suppression of polyglutamine‐induced cytotoxicity inSaccharomyces cerevisiaeby enhancement of mitochondrial biogenesis", The FASEB Journal, vol. 24, pp. 1431-1441, 2009. http://dx.doi.org/10.1096/fj.09-148601
- A. Solans, A. Zambrano, M. Rodríguez, and A. Barrientos, "Cytotoxicity of a mutant huntingtin fragment in yeast involves early alterations in mitochondrial OXPHOS complexes II and III", Human Molecular Genetics, vol. 15, pp. 3063-3081, 2006. http://dx.doi.org/10.1093/hmg/ddl248
- A.B. Meriin, X. Zhang, X. He, G.P. Newnam, Y.O. Chernoff, and M.Y. Sherman, "Huntingtin toxicity in yeast model depends on polyglutamine aggregation mediated by a prion-like protein Rnq1", The Journal of Cell Biology, vol. 157, pp. 997-1004, 2002. http://dx.doi.org/10.1083/jcb.200112104
- S. Willingham, T.F. Outeiro, M.J. DeVit, S.L. Lindquist, and P.J. Muchowski, "Yeast Genes That Enhance the Toxicity of a Mutant Huntingtin Fragment or α-Synuclein", Science, vol. 302, pp. 1769-1772, 2003. http://dx.doi.org/10.1126/science.1090389
- R.J. Braun, S. Büttner, J. Ring, G. Kroemer, and F. Madeo, "Nervous yeast: modeling neurotoxic cell death", Trends in Biochemical Sciences, vol. 35, pp. 135-144, 2010. http://dx.doi.org/10.1016/j.tibs.2009.10.005
- T.F. Outeiro, and F. Giorgini, "Yeast as a drug discovery platform in Huntington's and Parkinson's diseases", Biotechnology Journal, vol. 1, pp. 258-269, 2006. http://dx.doi.org/10.1002/biot.200500043
- S. Krobitsch, and S. Lindquist, "Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins", Proceedings of the National Academy of Sciences, vol. 97, pp. 1589-1594, 2000. http://dx.doi.org/10.1073/pnas.97.4.1589
- M.L. Duennwald, and S. Lindquist, "Impaired ERAD and ER stress are early and specific events in polyglutamine toxicity", Genes & Development, vol. 22, pp. 3308-3319, 2008. http://dx.doi.org/10.1101/gad.1673408
- S.L. Forsburg, and L. Guarente, "Identification and characterization of HAP4: a third component of the CCAAT-bound HAP2/HAP3 heteromer.", Genes & Development, vol. 3, pp. 1166-1178, 1989. http://dx.doi.org/10.1101/gad.3.8.1166
- S. Büttner, A. Bitto, J. Ring, M. Augsten, P. Zabrocki, T. Eisenberg, H. Jungwirth, S. Hutter, D. Carmona-Gutierrez, G. Kroemer, J. Winderickx, and F. Madeo, "Functional Mitochondria Are Required for α-Synuclein Toxicity in Aging Yeast", Journal of Biological Chemistry, vol. 283, pp. 7554-7560, 2008. http://dx.doi.org/10.1074/jbc.M708477200
- V. Longo, G. Shadel, M. Kaeberlein, and B. Kennedy, "Replicative and Chronological Aging in Saccharomyces cerevisiae", Cell Metabolism, vol. 16, pp. 18-31, 2012. http://dx.doi.org/10.1016/j.cmet.2012.06.002
- D.L. Smith, Jr, J.M. McClure, M. Matecic, and J.S. Smith, "Calorie restriction extends the chronological lifespan of Saccharomyces cerevisiae independently of the Sirtuins", Aging Cell, vol. 6, pp. 649-662, 2007. http://dx.doi.org/10.1111/j.1474-9726.2007.00326.x
- A. Ocampo, J. Liu, E. Schroeder, G. Shadel, and A. Barrientos, "Mitochondrial Respiratory Thresholds Regulate Yeast Chronological Life Span and its Extension by Caloric Restriction", Cell Metabolism, vol. 16, pp. 55-67, 2012. http://dx.doi.org/10.1016/j.cmet.2012.05.013
- A. Ocampo, and A. Barrientos, "From the Bakery to the Brain Business: Developing Inducible Yeast Models of Human Neurodegenerative Disorders", BioTechniques, vol. 45, 2008. http://dx.doi.org/10.2144/000112746
- J. Louvion, B. Havaux-Copf, and D. Picard, "Fusion of GAL4-VP16 to a steroid-binding domain provides a tool for gratuitous induction of galactose-responsive genes in yeast", Gene, vol. 131, pp. 129-134, 1993. http://dx.doi.org/10.1016/0378-1119(93)90681-r
- P.W. Piper, N.L. Harris, and M. MacLean, "Preadaptation to efficient respiratory maintenance is essential both for maximal longevity and the retention of replicative potential in chronologically ageing yeast", Mechanisms of Ageing and Development, vol. 127, pp. 733-740, 2006. http://dx.doi.org/10.1016/j.mad.2006.05.004
- M.H. Barros, B. Bandy, E.B. Tahara, and A.J. Kowaltowski, "Higher Respiratory Activity Decreases Mitochondrial Reactive Oxygen Release and Increases Life Span in Saccharomyces cerevisiae", Journal of Biological Chemistry, vol. 279, pp. 49883-49888, 2004. http://dx.doi.org/10.1074/jbc.m408918200
- C.R. Burtner, C.J. Murakami, B.K. Kennedy, and M. Kaeberlein, "A molecular mechanism of chronological aging in yeast", Cell Cycle, vol. 8, pp. 1256-1270, 2009. http://dx.doi.org/10.4161/cc.8.8.8287
- M. Matecic, D.L. Smith, X. Pan, N. Maqani, S. Bekiranov, J.D. Boeke, and J.S. Smith, "A Microarray-Based Genetic Screen for Yeast Chronological Aging Factors", PLoS Genetics, vol. 6, pp. e1000921, 2010. http://dx.doi.org/10.1371/journal.pgen.1000921
- M. Wei, P. Fabrizio, J. Hu, H. Ge, C. Cheng, L. Li, and V.D. Longo, "Life Span Extension by Calorie Restriction Depends on Rim15 and Transcription Factors Downstream of Ras/PKA, Tor, and Sch9", PLoS Genetics, vol. 4, pp. e13, 2008. http://dx.doi.org/10.1371/journal.pgen.0040013
- G.A. Oliveira, E.B. Tahara, A.K. Gombert, M.H. Barros, and A.J. Kowaltowski, "Increased aerobic metabolism is essential for the beneficial effects of caloric restriction on yeast life span", Journal of Bioenergetics and Biomembranes, vol. 40, 2008. http://dx.doi.org/10.1007/s10863-008-9159-5
- A. Ruetenik, and A. Barrientos, "Dietary restriction, mitochondrial function and aging: from yeast to humans", Biochimica et Biophysica Acta (BBA) - Bioenergetics, vol. 1847, pp. 1434-1447, 2015. http://dx.doi.org/10.1016/j.bbabio.2015.05.005
- M. Carlson, "Glucose repression in yeast", Current Opinion in Microbiology, vol. 2, pp. 202-207, 1999. http://dx.doi.org/10.1016/s1369-5274(99)80035-6
- M.J. Brauer, A.J. Saldanha, K. Dolinski, and D. Botstein, "Homeostatic Adjustment and Metabolic Remodeling in Glucose-limited Yeast Cultures", Molecular Biology of the Cell, vol. 16, pp. 2503-2517, 2005. http://dx.doi.org/10.1091/mbc.e04-11-0968
- G.G. Roberts, and A.P. Hudson, "Transcriptome profiling of Saccharomyces cerevisiae during a transition from fermentative to glycerol-based respiratory growth reveals extensive metabolic and structural remodeling", Molecular Genetics and Genomics, vol. 276, pp. 170-186, 2006. http://dx.doi.org/10.1007/s00438-006-0133-9
- M. Werner‐Washburne, E.L. Braun, M.E. Crawford, and V.M. Peck, "Stationary phase in Saccharomyces cerevisiae", Molecular Microbiology, vol. 19, pp. 1159-1166, 1996. http://dx.doi.org/10.1111/j.1365-2958.1996.tb02461.x
- A. Goldberg, S. Bourque, P. Kyryakov, T. Boukh-Viner, C. Gregg, A. Beach, M. Burstein, G. Machkalyan, V. Richard, S. Rampersad, and V. Titorenko, "A novel function of lipid droplets in regulating longevity", Biochemical Society Transactions, vol. 37, pp. 1050-1055, 2009. http://dx.doi.org/10.1042/BST0371050
- A. Ocampo, J. Liu, and A. Barrientos, "NAD+ salvage pathway proteins suppress proteotoxicity in yeast models of neurodegeneration by promoting the clearance of misfolded/oligomerized proteins", Human Molecular Genetics, vol. 22, pp. 1699-1708, 2013. http://dx.doi.org/10.1093/hmg/ddt016
- M. Rera, S. Bahadorani, J. Cho, C. Koehler, M. Ulgherait, J. Hur, W. Ansari, T. Lo, D. Jones, and D. Walker, "Modulation of Longevity and Tissue Homeostasis by the Drosophila PGC-1 Homolog", Cell Metabolism, vol. 14, pp. 623-634, 2011. http://dx.doi.org/10.1016/j.cmet.2011.09.013
- J.S. Steffan, L. Bodai, J. Pallos, M. Poelman, A. McCampbell, B.L. Apostol, A. Kazantsev, E. Schmidt, Y. Zhu, M. Greenwald, R. Kurokawa, D.E. Housman, G.R. Jackson, J.L. Marsh, and L.M. Thompson, "Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila", Nature, vol. 413, pp. 739-743, 2001. http://dx.doi.org/10.1038/35099568
- R.M. Ivatt, A. Sanchez-Martinez, V.K. Godena, S. Brown, E. Ziviani, and A.J. Whitworth, "Genome-wide RNAi screen identifies the Parkinson disease GWAS risk locusSREBF1as a regulator of mitophagy", Proceedings of the National Academy of Sciences, vol. 111, pp. 8494-8499, 2014. http://dx.doi.org/10.1073/pnas.1321207111
- C. Chen, Y. Hu, N.D. Udeshi, T.Y. Lau, F. Wirtz-Peitz, L. He, A.Y. Ting, S.A. Carr, and N. Perrimon, "Proteomic mapping in live Drosophila tissues using an engineered ascorbate peroxidase", Proceedings of the National Academy of Sciences, vol. 112, pp. 12093-12098, 2015. http://dx.doi.org/10.1073/pnas.1515623112
- S. Elmore, "Apoptosis: A Review of Programmed Cell Death", Toxicologic Pathology, vol. 35, pp. 495-516, 2007. http://dx.doi.org/10.1080/01926230701320337
- Z. Han, E.A. Hendrickson, T.A. Bremner, and J.H. Wyche, "A Sequential Two-Step Mechanism for the Production of the Mature p17:p12 Form of Caspase-3 in Vitro", Journal of Biological Chemistry, vol. 272, pp. 13432-13436, 1997. http://dx.doi.org/10.1074/jbc.272.20.13432
- T.F. Outeiro, and S. Lindquist, "Yeast Cells Provide Insight into Alpha-Synuclein Biology and Pathobiology", Science, vol. 302, pp. 1772-1775, 2003. http://dx.doi.org/10.1126/science.1090439
- A. Guedes, P. Ludovico, and B. Sampaio-Marques, "Caloric restriction alleviates alpha-synuclein toxicity in aged yeast cells by controlling the opposite roles of Tor1 and Sir2 on autophagy", Mechanisms of Ageing and Development, vol. 161, pp. 270-276, 2017. http://dx.doi.org/10.1016/j.mad.2016.04.006
- Y. Pan, E. Schroeder, A. Ocampo, A. Barrientos, and G. Shadel, "Regulation of Yeast Chronological Life Span by TORC1 via Adaptive Mitochondrial ROS Signaling", Cell Metabolism, vol. 13, pp. 668-678, 2011. http://dx.doi.org/10.1016/j.cmet.2011.03.018
- M. Chatenay-Lapointe, and G.S. Shadel, "Repression of Mitochondrial Translation, Respiration and a Metabolic Cycle-Regulated Gene, SLF1, by the Yeast Pumilio-Family Protein Puf3p", PLoS ONE, vol. 6, pp. e20441, 2011. http://dx.doi.org/10.1371/journal.pone.0020441
- W.J. Wolfgang, T.W. Miller, J.M. Webster, J.S. Huston, L.M. Thompson, J.L. Marsh, and A. Messer, "Suppression of Huntington's disease pathology in Drosophila by human single-chain Fv antibodies", Proceedings of the National Academy of Sciences, vol. 102, pp. 11563-11568, 2005. http://dx.doi.org/10.1073/pnas.0505321102
- P. Weydt, V.V. Pineda, A.E. Torrence, R.T. Libby, T.F. Satterfield, E. Lazarowski, M.L. Gilbert, G.J. Morton, T.K. Bammler, A.D. Strand, L. Cui, R.P. Beyer, C.N. Easley, A.C. Smith, D. Krainc, S. Luquet, I. Sweet, M.W. Schwartz, and A.R. La Spada, "Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1α in Huntington's disease neurodegeneration", Cell Metabolism, vol. 4, pp. 349-362, 2006. http://dx.doi.org/10.1016/j.cmet.2006.10.004
- P. Weydt, S.M. Soyal, C. Gellera, S. DiDonato, C. Weidinger, H. Oberkofler, G.B. Landwehrmeyer, and W. Patsch, "The gene coding for PGC-1α modifies age at onset in Huntington's Disease", Molecular Neurodegeneration, vol. 4, pp. 3, 2009. http://dx.doi.org/10.1186/1750-1326-4-3
- E. Taherzadeh-Fard, C. Saft, J. Andrich, S. Wieczorek, and L. Arning, "PGC-1alphaas modifier of onset age in Huntington disease", Molecular Neurodegeneration, vol. 4, 2009. http://dx.doi.org/10.1186/1750-1326-4-10
- P. Weydt, . , S.M. Soyal, G.B. Landwehrmeyer, and W. Patsch, "A single nucleotide polymorphism in the coding region of PGC-1α is a male-specific modifier of Huntington disease age-at-onset in a large European cohort", BMC Neurology, vol. 14, 2014. http://dx.doi.org/10.1186/1471-2377-14-1
- A.M. Myers, L.K. Pape, and A. Tzagoloff, "Mitochondrial protein synthesis is required for maintenance of intact mitochondrial genomes in Saccharomyces cerevisiae.", The EMBO journal, 1985. http://www.ncbi.nlm.nih.gov/pubmed/3905388
- P. Kyryakov, A. Beach, V.R. Richard, M.T. Burstein, A. Leonov, S. Levy, and V.I. Titorenko, "Caloric Restriction Extends Yeast Chronological Lifespan by Altering a Pattern of Age-Related Changes in Trehalose Concentration", Frontiers in Physiology, vol. 3, 2012. http://dx.doi.org/10.3389/fphys.2012.00256
- N. Guaragnella, M. Ždralević, P. Lattanzio, D. Marzulli, T. Pracheil, Z. Liu, S. Passarella, E. Marra, and S. Giannattasio, "Yeast growth in raffinose results in resistance to acetic-acid induced programmed cell death mostly due to the activation of the mitochondrial retrograde pathway", Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, vol. 1833, pp. 2765-2774, 2013. http://dx.doi.org/10.1016/j.bbamcr.2013.07.017
- A. Ocampo, and A. Barrientos, "Quick and reliable assessment of chronological life span in yeast cell populations by flow cytometry", Mechanisms of Ageing and Development, vol. 132, pp. 315-323, 2011. http://dx.doi.org/10.1016/j.mad.2011.06.007
- A. Barrientos, "In vivo and in organello assessment of OXPHOS activities", Methods, vol. 26, pp. 307-316, 2002. http://dx.doi.org/10.1016/s1046-2023(02)00036-1
- S.K. Tiefenböck, C. Baltzer, N.A. Egli, and C. Frei, "The Drosophila PGC-1 homologue Spargel coordinates mitochondrial activity to insulin signalling", The EMBO Journal, vol. 29, pp. 171-183, 2009. http://dx.doi.org/10.1038/emboj.2009.330
- Y.O. Ali, W. Escala, K. Ruan, and R.G. Zhai, "Assaying Locomotor, Learning, and Memory Deficits in <em>Drosophila</em> Models of Neurodegeneration", Journal of Visualized Experiments, 2011. http://dx.doi.org/10.3791/2504
- K. Ruan, Y. Zhu, C. Li, J.M. Brazill, and R.G. Zhai, "Alternative splicing of Drosophila Nmnat functions as a switch to enhance neuroprotection under stress", Nature Communications, vol. 6, 2015. http://dx.doi.org/10.1038/ncomms10057
SUPPLEMENTAL INFORMATION
 Download Supplemental Information
Download Supplemental Information
ACKNOWLEDGMENTS
This work is supported by grants from the National Institutes of Health (NIH) R01 GM071775, GM105781 and GM112179 (to A.B.) and 2R56NS064269 (to R.G.Z), and of the US Department of the Army (ARO # #65594-LS to A.B.).
COPYRIGHT
© 2016

Attenuation of polyglutamine-induced toxicity by enhancement of mitochondrial OXPHOS in yeast and fly models of aging by Andrea L. Ruetenik et al. is licensed under a Creative Commons Attribution 4.0 International License.








